 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn SO(3)-equivariant reciprocal-space neural potential for long-range interactions
Mar 19, 2026Long-range electrostatic and polarization interactions play a central role in molecular and condensed-phase systems, yet remain fundamentally incompatible with locality-based machine-learning interatomic potentials. Although modern SO(3)-equivariant neural potentials achieve high accuracy for short-range chemistry, they cannot represent the anisotropic, slowly decaying multipolar correlations governing realistic materials, while existing long-range extensions either break SO(3) equivariance or fail to maintain energy-force consistency. Here we introduce EquiEwald, a unified neural interatomic potential that embeds an Ewald-inspired reciprocal-space formulation within an irreducible SO(3)-equivariant framework. By performing equivariant message passing in reciprocal space through learned equivariant k-space filters and an equivariant inverse transform, EquiEwald captures anisotropic, tensorial long-range correlations without sacrificing physical consistency. Across periodic and aperiodic benchmarks, EquiEwald captures long-range electrostatic behavior consistent with ab initio reference data and consistently improves energy and force accuracy, data efficiency, and long-range extrapolation. These results establish EquiEwald as a physically principled paradigm for long-range-capable machine-learning interatomic potentials.
Discovery of Interpretable Physical Laws in Materials via Language-Model-Guided Symbolic Regression
Feb 26, 2026Discovering interpretable physical laws from high-dimensional data is a fundamental challenge in scientific research. Traditional methods, such as symbolic regression, often produce complex, unphysical formulas when searching a vast space of possible forms. We introduce a framework that guides the search process by leveraging the embedded scientific knowledge of large language models, enabling efficient identification of physical laws in the data. We validate our approach by modeling key properties of perovskite materials. Our method mitigates the combinatorial explosion commonly encountered in traditional symbolic regression, reducing the effective search space by a factor of approximately $10^5$. A set of novel formulas for bulk modulus, band gap, and oxygen evolution reaction activity are identified, which not only provide meaningful physical insights but also outperform previous formulas in accuracy and simplicity.
Equivariant Evidential Deep Learning for Interatomic Potentials
Feb 11, 2026Uncertainty quantification (UQ) is critical for assessing the reliability of machine learning interatomic potentials (MLIPs) in molecular dynamics (MD) simulations, identifying extrapolation regimes and enabling uncertainty-aware workflows such as active learning for training dataset construction. Existing UQ approaches for MLIPs are often limited by high computational cost or suboptimal performance. Evidential deep learning (EDL) provides a theoretically grounded single-model alternative that determines both aleatoric and epistemic uncertainty in a single forward pass. However, extending evidential formulations from scalar targets to vector-valued quantities such as atomic forces introduces substantial challenges, particularly in maintaining statistical self-consistency under rotational transformations. To address this, we propose \textit{Equivariant Evidential Deep Learning for Interatomic Potentials} ($\text{e}^2$IP), a backbone-agnostic framework that models atomic forces and their uncertainty jointly by representing uncertainty as a full $3\times3$ symmetric positive definite covariance tensor that transforms equivariantly under rotations. Experiments on diverse molecular benchmarks show that $\text{e}^2$IP provides a stronger accuracy-efficiency-reliability balance than the non-equivariant evidential baseline and the widely used ensemble method. It also achieves better data efficiency through the fully equivariant architecture while retaining single-model inference efficiency.
An Agentic Framework for Autonomous Materials Computation
Dec 22, 2025



Large Language Models (LLMs) have emerged as powerful tools for accelerating scientific discovery, yet their static knowledge and hallucination issues hinder autonomous research applications. Recent advances integrate LLMs into agentic frameworks, enabling retrieval, reasoning, and tool use for complex scientific workflows. Here, we present a domain-specialized agent designed for reliable automation of first-principles materials computations. By embedding domain expertise, the agent ensures physically coherent multi-step workflows and consistently selects convergent, well-posed parameters, thereby enabling reliable end-to-end computational execution. A new benchmark of diverse computational tasks demonstrates that our system significantly outperforms standalone LLMs in both accuracy and robustness. This work establishes a verifiable foundation for autonomous computational experimentation and represents a key step toward fully automated scientific discovery.
Probing Scientific General Intelligence of LLMs with Scientist-Aligned Workflows
Dec 18, 2025Despite advances in scientific AI, a coherent framework for Scientific General Intelligence (SGI)-the ability to autonomously conceive, investigate, and reason across scientific domains-remains lacking. We present an operational SGI definition grounded in the Practical Inquiry Model (PIM: Deliberation, Conception, Action, Perception) and operationalize it via four scientist-aligned tasks: deep research, idea generation, dry/wet experiments, and experimental reasoning. SGI-Bench comprises over 1,000 expert-curated, cross-disciplinary samples inspired by Science's 125 Big Questions, enabling systematic evaluation of state-of-the-art LLMs. Results reveal gaps: low exact match (10--20%) in deep research despite step-level alignment; ideas lacking feasibility and detail; high code executability but low execution result accuracy in dry experiments; low sequence fidelity in wet protocols; and persistent multimodal comparative-reasoning challenges. We further introduce Test-Time Reinforcement Learning (TTRL), which optimizes retrieval-augmented novelty rewards at inference, enhancing hypothesis novelty without reference answer. Together, our PIM-grounded definition, workflow-centric benchmark, and empirical insights establish a foundation for AI systems that genuinely participate in scientific discovery.
ATLAS: A High-Difficulty, Multidisciplinary Benchmark for Frontier Scientific Reasoning
Nov 18, 2025The rapid advancement of Large Language Models (LLMs) has led to performance saturation on many established benchmarks, questioning their ability to distinguish frontier models. Concurrently, existing high-difficulty benchmarks often suffer from narrow disciplinary focus, oversimplified answer formats, and vulnerability to data contamination, creating a fidelity gap with real-world scientific inquiry. To address these challenges, we introduce ATLAS (AGI-Oriented Testbed for Logical Application in Science), a large-scale, high-difficulty, and cross-disciplinary evaluation suite composed of approximately 800 original problems. Developed by domain experts (PhD-level and above), ATLAS spans seven core scientific fields: mathematics, physics, chemistry, biology, computer science, earth science, and materials science. Its key features include: (1) High Originality and Contamination Resistance, with all questions newly created or substantially adapted to prevent test data leakage; (2) Cross-Disciplinary Focus, designed to assess models' ability to integrate knowledge and reason across scientific domains; (3) High-Fidelity Answers, prioritizing complex, open-ended answers involving multi-step reasoning and LaTeX-formatted expressions over simple multiple-choice questions; and (4) Rigorous Quality Control, employing a multi-stage process of expert peer review and adversarial testing to ensure question difficulty, scientific value, and correctness. We also propose a robust evaluation paradigm using a panel of LLM judges for automated, nuanced assessment of complex answers. Preliminary results on leading models demonstrate ATLAS's effectiveness in differentiating their advanced scientific reasoning capabilities. We plan to develop ATLAS into a long-term, open, community-driven platform to provide a reliable "ruler" for progress toward Artificial General Intelligence.
ChemBOMAS: Accelerated BO in Chemistry with LLM-Enhanced Multi-Agent System
Sep 10, 2025The efficiency of Bayesian optimization (BO) in chemistry is often hindered by sparse experimental data and complex reaction mechanisms. To overcome these limitations, we introduce ChemBOMAS, a new framework named LLM-Enhanced Multi-Agent System for accelerating BO in chemistry. ChemBOMAS's optimization process is enhanced by LLMs and synergistically employs two strategies: knowledge-driven coarse-grained optimization and data-driven fine-grained optimization. First, in the knowledge-driven coarse-grained optimization stage, LLMs intelligently decompose the vast search space by reasoning over existing chemical knowledge to identify promising candidate regions. Subsequently, in the data-driven fine-grained optimization stage, LLMs enhance the BO process within these candidate regions by generating pseudo-data points, thereby improving data utilization efficiency and accelerating convergence. Benchmark evaluations** further confirm that ChemBOMAS significantly enhances optimization effectiveness and efficiency compared to various BO algorithms. Importantly, the practical utility of ChemBOMAS was validated through wet-lab experiments conducted under pharmaceutical industry protocols, targeting conditional optimization for a previously unreported and challenging chemical reaction. In the wet experiment, ChemBOMAS achieved an optimal objective value of 96%. This was substantially higher than the 15% achieved by domain experts. This real-world success, together with strong performance on benchmark evaluations, highlights ChemBOMAS as a powerful tool to accelerate chemical discovery.
CMPhysBench: A Benchmark for Evaluating Large Language Models in Condensed Matter Physics
Aug 25, 2025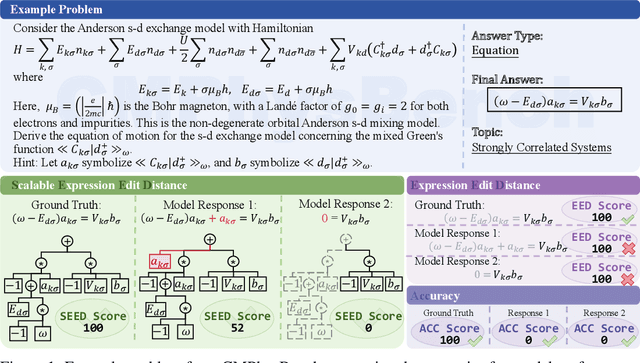
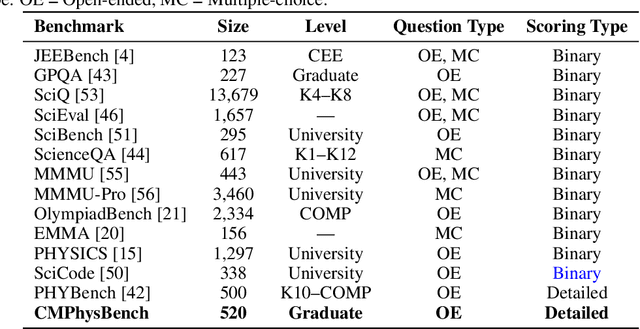
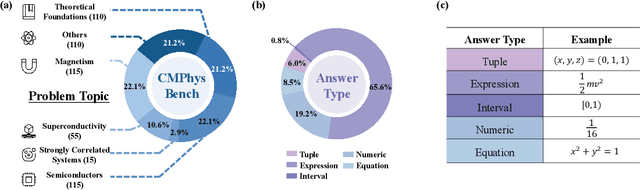
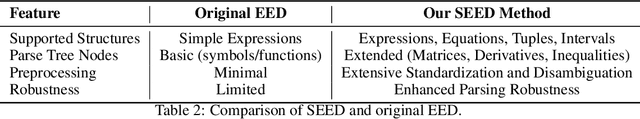
We introduce CMPhysBench, designed to assess the proficiency of Large Language Models (LLMs) in Condensed Matter Physics, as a novel Benchmark. CMPhysBench is composed of more than 520 graduate-level meticulously curated questions covering both representative subfields and foundational theoretical frameworks of condensed matter physics, such as magnetism, superconductivity, strongly correlated systems, etc. To ensure a deep understanding of the problem-solving process,we focus exclusively on calculation problems, requiring LLMs to independently generate comprehensive solutions. Meanwhile, leveraging tree-based representations of expressions, we introduce the Scalable Expression Edit Distance (SEED) score, which provides fine-grained (non-binary) partial credit and yields a more accurate assessment of similarity between prediction and ground-truth. Our results show that even the best models, Grok-4, reach only 36 average SEED score and 28% accuracy on CMPhysBench, underscoring a significant capability gap, especially for this practical and frontier domain relative to traditional physics. The code anddataset are publicly available at https://github.com/CMPhysBench/CMPhysBench.
Iterative Pretraining Framework for Interatomic Potentials
Jul 27, 2025Machine learning interatomic potentials (MLIPs) enable efficient molecular dynamics (MD) simulations with ab initio accuracy and have been applied across various domains in physical science. However, their performance often relies on large-scale labeled training data. While existing pretraining strategies can improve model performance, they often suffer from a mismatch between the objectives of pretraining and downstream tasks or rely on extensive labeled datasets and increasingly complex architectures to achieve broad generalization. To address these challenges, we propose Iterative Pretraining for Interatomic Potentials (IPIP), a framework designed to iteratively improve the predictive performance of MLIP models. IPIP incorporates a forgetting mechanism to prevent iterative training from converging to suboptimal local minima. Unlike general-purpose foundation models, which frequently underperform on specialized tasks due to a trade-off between generality and system-specific accuracy, IPIP achieves higher accuracy and efficiency using lightweight architectures. Compared to general-purpose force fields, this approach achieves over 80% reduction in prediction error and up to 4x speedup in the challenging Mo-S-O system, enabling fast and accurate simulations.
ChemVLM: Exploring the Power of Multimodal Large Language Models in Chemistry Area
Aug 16, 2024



Large Language Models (LLMs) have achieved remarkable success and have been applied across various scientific fields, including chemistry. However, many chemical tasks require the processing of visual information, which cannot be successfully handled by existing chemical LLMs. This brings a growing need for models capable of integrating multimodal information in the chemical domain. In this paper, we introduce \textbf{ChemVLM}, an open-source chemical multimodal large language model specifically designed for chemical applications. ChemVLM is trained on a carefully curated bilingual multimodal dataset that enhances its ability to understand both textual and visual chemical information, including molecular structures, reactions, and chemistry examination questions. We develop three datasets for comprehensive evaluation, tailored to Chemical Optical Character Recognition (OCR), Multimodal Chemical Reasoning (MMCR), and Multimodal Molecule Understanding tasks. We benchmark ChemVLM against a range of open-source and proprietary multimodal large language models on various tasks. Experimental results demonstrate that ChemVLM achieves competitive performance across all evaluated tasks. Our model can be found at https://huggingface.co/AI4Chem/ChemVLM-26B.