 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEDT: An Efficient Diffusion Transformer Framework Inspired by Human-like Sketching
Oct 31, 2024Transformer-based Diffusion Probabilistic Models (DPMs) have shown more potential than CNN-based DPMs, yet their extensive computational requirements hinder widespread practical applications. To reduce the computation budget of transformer-based DPMs, this work proposes the Efficient Diffusion Transformer (EDT) framework. The framework includes a lightweight-design diffusion model architecture, and a training-free Attention Modulation Matrix and its alternation arrangement in EDT inspired by human-like sketching. Additionally, we propose a token relation-enhanced masking training strategy tailored explicitly for EDT to augment its token relation learning capability. Our extensive experiments demonstrate the efficacy of EDT. The EDT framework reduces training and inference costs and surpasses existing transformer-based diffusion models in image synthesis performance, thereby achieving a significant overall enhancement. With lower FID, EDT-S, EDT-B, and EDT-XL attained speed-ups of 3.93x, 2.84x, and 1.92x respectively in the training phase, and 2.29x, 2.29x, and 2.22x respectively in inference, compared to the corresponding sizes of MDTv2. The source code is released at https://github.com/xinwangChen/EDT.
Structure Language Models for Protein Conformation Generation
Oct 24, 2024



Proteins adopt multiple structural conformations to perform their diverse biological functions, and understanding these conformations is crucial for advancing drug discovery. Traditional physics-based simulation methods often struggle with sampling equilibrium conformations and are computationally expensive. Recently, deep generative models have shown promise in generating protein conformations as a more efficient alternative. However, these methods predominantly rely on the diffusion process within a 3D geometric space, which typically centers around the vicinity of metastable states and is often inefficient in terms of runtime. In this paper, we introduce Structure Language Modeling (SLM) as a novel framework for efficient protein conformation generation. Specifically, the protein structures are first encoded into a compact latent space using a discrete variational auto-encoder, followed by conditional language modeling that effectively captures sequence-specific conformation distributions. This enables a more efficient and interpretable exploration of diverse ensemble modes compared to existing methods. Based on this general framework, we instantiate SLM with various popular LM architectures as well as proposing the ESMDiff, a novel BERT-like structure language model fine-tuned from ESM3 with masked diffusion. We verify our approach in various scenarios, including the equilibrium dynamics of BPTI, conformational change pairs, and intrinsically disordered proteins. SLM provides a highly efficient solution, offering a 20-100x speedup than existing methods in generating diverse conformations, shedding light on promising avenues for future research.
REGNet V2: End-to-End REgion-based Grasp Detection Network for Grippers of Different Sizes in Point Clouds
Oct 12, 2024
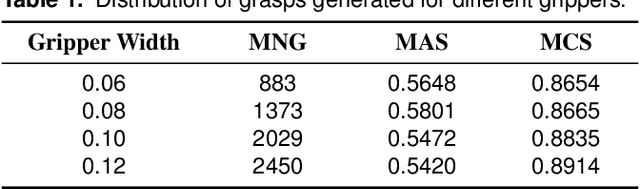
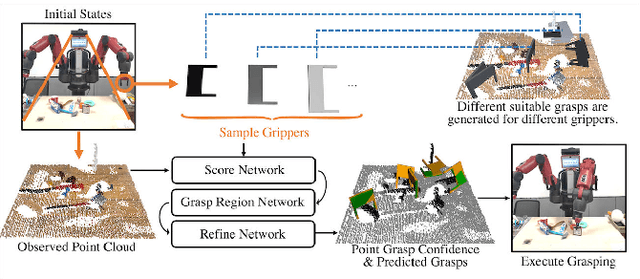
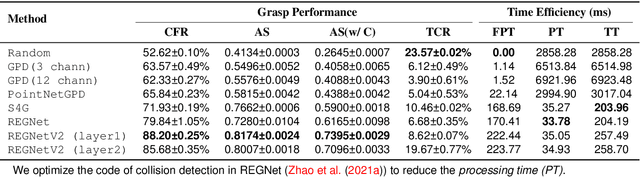
Grasping has been a crucial but challenging problem in robotics for many years. One of the most important challenges is how to make grasping generalizable and robust to novel objects as well as grippers in unstructured environments. We present \regnet, a robotic grasping system that can adapt to different parallel jaws to grasp diversified objects. To support different grippers, \regnet embeds the gripper parameters into point clouds, based on which it predicts suitable grasp configurations. It includes three components: Score Network (SN), Grasp Region Network (GRN), and Refine Network (RN). In the first stage, SN is used to filter suitable points for grasping by grasp confidence scores. In the second stage, based on the selected points, GRN generates a set of grasp proposals. Finally, RN refines the grasp proposals for more accurate and robust predictions. We devise an analytic policy to choose the optimal grasp to be executed from the predicted grasp set. To train \regnet, we construct a large-scale grasp dataset containing collision-free grasp configurations using different parallel-jaw grippers. The experimental results demonstrate that \regnet with the analytic policy achieves the highest success rate of $74.98\%$ in real-world clutter scenes with $20$ objects, significantly outperforming several state-of-the-art methods, including GPD, PointNetGPD, and S4G. The code and dataset are available at https://github.com/zhaobinglei/REGNet-V2.
Discrete Policy: Learning Disentangled Action Space for Multi-Task Robotic Manipulation
Sep 27, 2024



Learning visuomotor policy for multi-task robotic manipulation has been a long-standing challenge for the robotics community. The difficulty lies in the diversity of action space: typically, a goal can be accomplished in multiple ways, resulting in a multimodal action distribution for a single task. The complexity of action distribution escalates as the number of tasks increases. In this work, we propose \textbf{Discrete Policy}, a robot learning method for training universal agents capable of multi-task manipulation skills. Discrete Policy employs vector quantization to map action sequences into a discrete latent space, facilitating the learning of task-specific codes. These codes are then reconstructed into the action space conditioned on observations and language instruction. We evaluate our method on both simulation and multiple real-world embodiments, including both single-arm and bimanual robot settings. We demonstrate that our proposed Discrete Policy outperforms a well-established Diffusion Policy baseline and many state-of-the-art approaches, including ACT, Octo, and OpenVLA. For example, in a real-world multi-task training setting with five tasks, Discrete Policy achieves an average success rate that is 26\% higher than Diffusion Policy and 15\% higher than OpenVLA. As the number of tasks increases to 12, the performance gap between Discrete Policy and Diffusion Policy widens to 32.5\%, further showcasing the advantages of our approach. Our work empirically demonstrates that learning multi-task policies within the latent space is a vital step toward achieving general-purpose agents.
Scaling Diffusion Policy in Transformer to 1 Billion Parameters for Robotic Manipulation
Sep 22, 2024



Diffusion Policy is a powerful technique tool for learning end-to-end visuomotor robot control. It is expected that Diffusion Policy possesses scalability, a key attribute for deep neural networks, typically suggesting that increasing model size would lead to enhanced performance. However, our observations indicate that Diffusion Policy in transformer architecture (\DP) struggles to scale effectively; even minor additions of layers can deteriorate training outcomes. To address this issue, we introduce Scalable Diffusion Transformer Policy for visuomotor learning. Our proposed method, namely \textbf{\methodname}, introduces two modules that improve the training dynamic of Diffusion Policy and allow the network to better handle multimodal action distribution. First, we identify that \DP~suffers from large gradient issues, making the optimization of Diffusion Policy unstable. To resolve this issue, we factorize the feature embedding of observation into multiple affine layers, and integrate it into the transformer blocks. Additionally, our utilize non-causal attention which allows the policy network to \enquote{see} future actions during prediction, helping to reduce compounding errors. We demonstrate that our proposed method successfully scales the Diffusion Policy from 10 million to 1 billion parameters. This new model, named \methodname, can effectively scale up the model size with improved performance and generalization. We benchmark \methodname~across 50 different tasks from MetaWorld and find that our largest \methodname~outperforms \DP~with an average improvement of 21.6\%. Across 7 real-world robot tasks, our ScaleDP demonstrates an average improvement of 36.25\% over DP-T on four single-arm tasks and 75\% on three bimanual tasks. We believe our work paves the way for scaling up models for visuomotor learning. The project page is available at scaling-diffusion-policy.github.io.
Are Heterophily-Specific GNNs and Homophily Metrics Really Effective? Evaluation Pitfalls and New Benchmarks
Sep 09, 2024



Over the past decade, Graph Neural Networks (GNNs) have achieved great success on machine learning tasks with relational data. However, recent studies have found that heterophily can cause significant performance degradation of GNNs, especially on node-level tasks. Numerous heterophilic benchmark datasets have been put forward to validate the efficacy of heterophily-specific GNNs and various homophily metrics have been designed to help people recognize these malignant datasets. Nevertheless, there still exist multiple pitfalls that severely hinder the proper evaluation of new models and metrics. In this paper, we point out three most serious pitfalls: 1) a lack of hyperparameter tuning; 2) insufficient model evaluation on the real challenging heterophilic datasets; 3) missing quantitative evaluation benchmark for homophily metrics on synthetic graphs. To overcome these challenges, we first train and fine-tune baseline models on $27$ most widely used benchmark datasets, categorize them into three distinct groups: malignant, benign and ambiguous heterophilic datasets, and identify the real challenging subsets of tasks. To our best knowledge, we are the first to propose such taxonomy. Then, we re-evaluate $10$ heterophily-specific state-of-the-arts (SOTA) GNNs with fine-tuned hyperparameters on different groups of heterophilic datasets. Based on the model performance, we reassess their effectiveness on addressing heterophily challenge. At last, we evaluate $11$ popular homophily metrics on synthetic graphs with three different generation approaches. To compare the metrics strictly, we propose the first quantitative evaluation method based on Fr\'echet distance.
DexDiff: Towards Extrinsic Dexterity Manipulation of Ungraspable Objects in Unrestricted Environments
Sep 09, 2024



Grasping large and flat objects (e.g. a book or a pan) is often regarded as an ungraspable task, which poses significant challenges due to the unreachable grasping poses. Previous works leverage Extrinsic Dexterity like walls or table edges to grasp such objects. However, they are limited to task-specific policies and lack task planning to find pre-grasp conditions. This makes it difficult to adapt to various environments and extrinsic dexterity constraints. Therefore, we present DexDiff, a robust robotic manipulation method for long-horizon planning with extrinsic dexterity. Specifically, we utilize a vision-language model (VLM) to perceive the environmental state and generate high-level task plans, followed by a goal-conditioned action diffusion (GCAD) model to predict the sequence of low-level actions. This model learns the low-level policy from offline data with the cumulative reward guided by high-level planning as the goal condition, which allows for improved prediction of robot actions. Experimental results demonstrate that our method not only effectively performs ungraspable tasks but also generalizes to previously unseen objects. It outperforms baselines by a 47% higher success rate in simulation and facilitates efficient deployment and manipulation in real-world scenarios.
Cell-ontology guided transcriptome foundation model
Aug 22, 2024



Transcriptome foundation models TFMs hold great promises of deciphering the transcriptomic language that dictate diverse cell functions by self-supervised learning on large-scale single-cell gene expression data, and ultimately unraveling the complex mechanisms of human diseases. However, current TFMs treat cells as independent samples and ignore the taxonomic relationships between cell types, which are available in cell ontology graphs. We argue that effectively leveraging this ontology information during the TFM pre-training can improve learning biologically meaningful gene co-expression patterns while preserving TFM as a general purpose foundation model for downstream zero-shot and fine-tuning tasks. To this end, we present \textbf{s}ingle \textbf{c}ell, \textbf{Cell}-\textbf{o}ntology guided TFM scCello. We introduce cell-type coherence loss and ontology alignment loss, which are minimized along with the masked gene expression prediction loss during the pre-training. The novel loss component guide scCello to learn the cell-type-specific representation and the structural relation between cell types from the cell ontology graph, respectively. We pre-trained scCello on 22 million cells from CellxGene database leveraging their cell-type labels mapped to the cell ontology graph from Open Biological and Biomedical Ontology Foundry. Our TFM demonstrates competitive generalization and transferability performance over the existing TFMs on biologically important tasks including identifying novel cell types of unseen cells, prediction of cell-type-specific marker genes, and cancer drug responses.
Moment&Cross: Next-Generation Real-Time Cross-Domain CTR Prediction for Live-Streaming Recommendation at Kuaishou
Aug 11, 2024



Kuaishou, is one of the largest short-video and live-streaming platform, compared with short-video recommendations, live-streaming recommendation is more complex because of: (1) temporarily-alive to distribution, (2) user may watch for a long time with feedback delay, (3) content is unpredictable and changes over time. Actually, even if a user is interested in the live-streaming author, it still may be an negative watching (e.g., short-view < 3s) since the real-time content is not attractive enough. Therefore, for live-streaming recommendation, there exists a challenging task: how do we recommend the live-streaming at right moment for users? Additionally, our platform's major exposure content is short short-video, and the amount of exposed short-video is 9x more than exposed live-streaming. Thus users will leave more behaviors on short-videos, which leads to a serious data imbalance problem making the live-streaming data could not fully reflect user interests. In such case, there raises another challenging task: how do we utilize users' short-video behaviors to make live-streaming recommendation better?
The Heterophilic Graph Learning Handbook: Benchmarks, Models, Theoretical Analysis, Applications and Challenges
Jul 12, 2024



Homophily principle, \ie{} nodes with the same labels or similar attributes are more likely to be connected, has been commonly believed to be the main reason for the superiority of Graph Neural Networks (GNNs) over traditional Neural Networks (NNs) on graph-structured data, especially on node-level tasks. However, recent work has identified a non-trivial set of datasets where GNN's performance compared to the NN's is not satisfactory. Heterophily, i.e. low homophily, has been considered the main cause of this empirical observation. People have begun to revisit and re-evaluate most existing graph models, including graph transformer and its variants, in the heterophily scenario across various kinds of graphs, e.g. heterogeneous graphs, temporal graphs and hypergraphs. Moreover, numerous graph-related applications are found to be closely related to the heterophily problem. In the past few years, considerable effort has been devoted to studying and addressing the heterophily issue. In this survey, we provide a comprehensive review of the latest progress on heterophilic graph learning, including an extensive summary of benchmark datasets and evaluation of homophily metrics on synthetic graphs, meticulous classification of the most updated supervised and unsupervised learning methods, thorough digestion of the theoretical analysis on homophily/heterophily, and broad exploration of the heterophily-related applications. Notably, through detailed experiments, we are the first to categorize benchmark heterophilic datasets into three sub-categories: malignant, benign and ambiguous heterophily. Malignant and ambiguous datasets are identified as the real challenging datasets to test the effectiveness of new models on the heterophily challenge. Finally, we propose several challenges and future directions for heterophilic graph representation learning.