 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCo$^{3}$Gesture: Towards Coherent Concurrent Co-speech 3D Gesture Generation with Interactive Diffusion
May 03, 2025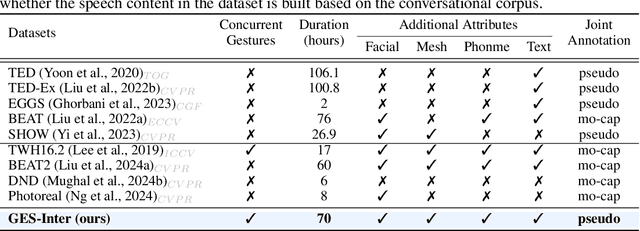

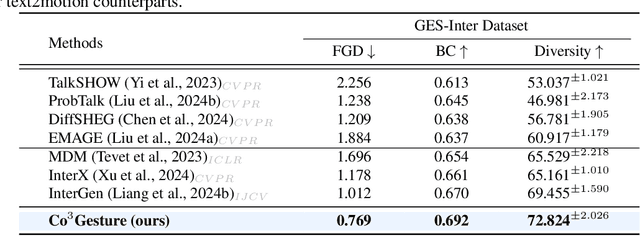
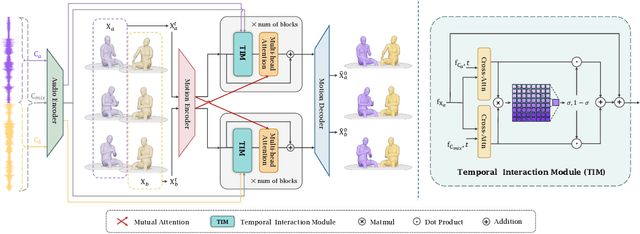
Generating gestures from human speech has gained tremendous progress in animating virtual avatars. While the existing methods enable synthesizing gestures cooperated by individual self-talking, they overlook the practicality of concurrent gesture modeling with two-person interactive conversations. Moreover, the lack of high-quality datasets with concurrent co-speech gestures also limits handling this issue. To fulfill this goal, we first construct a large-scale concurrent co-speech gesture dataset that contains more than 7M frames for diverse two-person interactive posture sequences, dubbed GES-Inter. Additionally, we propose Co$^3$Gesture, a novel framework that enables coherent concurrent co-speech gesture synthesis including two-person interactive movements. Considering the asymmetric body dynamics of two speakers, our framework is built upon two cooperative generation branches conditioned on separated speaker audio. Specifically, to enhance the coordination of human postures with respect to corresponding speaker audios while interacting with the conversational partner, we present a Temporal Interaction Module (TIM). TIM can effectively model the temporal association representation between two speakers' gesture sequences as interaction guidance and fuse it into the concurrent gesture generation. Then, we devise a mutual attention mechanism to further holistically boost learning dependencies of interacted concurrent motions, thereby enabling us to generate vivid and coherent gestures. Extensive experiments demonstrate that our method outperforms the state-of-the-art models on our newly collected GES-Inter dataset. The dataset and source code are publicly available at \href{https://mattie-e.github.io/Co3/}{\textit{https://mattie-e.github.io/Co3/}}.
Benchmarking Multi-National Value Alignment for Large Language Models
Apr 19, 2025



Do Large Language Models (LLMs) hold positions that conflict with your country's values? Occasionally they do! However, existing works primarily focus on ethical reviews, failing to capture the diversity of national values, which encompass broader policy, legal, and moral considerations. Furthermore, current benchmarks that rely on spectrum tests using manually designed questionnaires are not easily scalable. To address these limitations, we introduce NaVAB, a comprehensive benchmark to evaluate the alignment of LLMs with the values of five major nations: China, the United States, the United Kingdom, France, and Germany. NaVAB implements a national value extraction pipeline to efficiently construct value assessment datasets. Specifically, we propose a modeling procedure with instruction tagging to process raw data sources, a screening process to filter value-related topics and a generation process with a Conflict Reduction mechanism to filter non-conflicting values.We conduct extensive experiments on various LLMs across countries, and the results provide insights into assisting in the identification of misaligned scenarios. Moreover, we demonstrate that NaVAB can be combined with alignment techniques to effectively reduce value concerns by aligning LLMs' values with the target country.
ThinkPatterns-21k: A Systematic Study on the Impact of Thinking Patterns in LLMs
Mar 17, 2025Large language models (LLMs) have demonstrated enhanced performance through the \textit{Thinking then Responding} paradigm, where models generate internal thoughts before final responses (aka, System 2 thinking). However, existing research lacks a systematic understanding of the mechanisms underlying how thinking patterns affect performance across model sizes. In this work, we conduct a comprehensive analysis of the impact of various thinking types on model performance and introduce ThinkPatterns-21k, a curated dataset comprising 21k instruction-response pairs (QA) collected from existing instruction-following datasets with five thinking types. For each pair, we augment it with five distinct internal thinking patterns: one unstructured thinking (monologue) and four structured variants (decomposition, self-ask, self-debate and self-critic), while maintaining the same instruction and response. Through extensive evaluation across different model sizes (3B-32B parameters), we have two key findings: (1) smaller models (<30B parameters) can benefit from most of structured thinking patterns, while larger models (32B) with structured thinking like decomposition would degrade performance and (2) unstructured monologue demonstrates broad effectiveness across different model sizes. Finally, we released all of our datasets, checkpoints, training logs of diverse thinking patterns to reproducibility, aiming to facilitate further research in this direction.
AudioX: Diffusion Transformer for Anything-to-Audio Generation
Mar 13, 2025Audio and music generation have emerged as crucial tasks in many applications, yet existing approaches face significant limitations: they operate in isolation without unified capabilities across modalities, suffer from scarce high-quality, multi-modal training data, and struggle to effectively integrate diverse inputs. In this work, we propose AudioX, a unified Diffusion Transformer model for Anything-to-Audio and Music Generation. Unlike previous domain-specific models, AudioX can generate both general audio and music with high quality, while offering flexible natural language control and seamless processing of various modalities including text, video, image, music, and audio. Its key innovation is a multi-modal masked training strategy that masks inputs across modalities and forces the model to learn from masked inputs, yielding robust and unified cross-modal representations. To address data scarcity, we curate two comprehensive datasets: vggsound-caps with 190K audio captions based on the VGGSound dataset, and V2M-caps with 6 million music captions derived from the V2M dataset. Extensive experiments demonstrate that AudioX not only matches or outperforms state-of-the-art specialized models, but also offers remarkable versatility in handling diverse input modalities and generation tasks within a unified architecture. The code and datasets will be available at https://zeyuet.github.io/AudioX/
YuE: Scaling Open Foundation Models for Long-Form Music Generation
Mar 11, 2025We tackle the task of long-form music generation--particularly the challenging \textbf{lyrics-to-song} problem--by introducing YuE, a family of open foundation models based on the LLaMA2 architecture. Specifically, YuE scales to trillions of tokens and generates up to five minutes of music while maintaining lyrical alignment, coherent musical structure, and engaging vocal melodies with appropriate accompaniment. It achieves this through (1) track-decoupled next-token prediction to overcome dense mixture signals, (2) structural progressive conditioning for long-context lyrical alignment, and (3) a multitask, multiphase pre-training recipe to converge and generalize. In addition, we redesign the in-context learning technique for music generation, enabling versatile style transfer (e.g., converting Japanese city pop into an English rap while preserving the original accompaniment) and bidirectional generation. Through extensive evaluation, we demonstrate that YuE matches or even surpasses some of the proprietary systems in musicality and vocal agility. In addition, fine-tuning YuE enables additional controls and enhanced support for tail languages. Furthermore, beyond generation, we show that YuE's learned representations can perform well on music understanding tasks, where the results of YuE match or exceed state-of-the-art methods on the MARBLE benchmark. Keywords: lyrics2song, song generation, long-form, foundation model, music generation
Spark-TTS: An Efficient LLM-Based Text-to-Speech Model with Single-Stream Decoupled Speech Tokens
Mar 03, 2025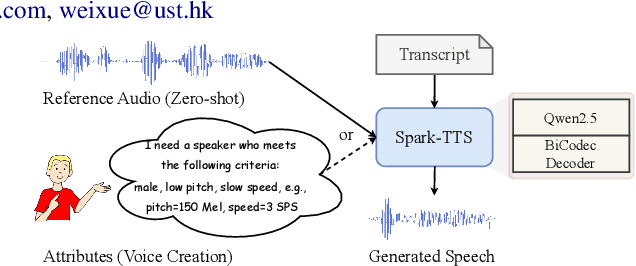



Recent advancements in large language models (LLMs) have driven significant progress in zero-shot text-to-speech (TTS) synthesis. However, existing foundation models rely on multi-stage processing or complex architectures for predicting multiple codebooks, limiting efficiency and integration flexibility. To overcome these challenges, we introduce Spark-TTS, a novel system powered by BiCodec, a single-stream speech codec that decomposes speech into two complementary token types: low-bitrate semantic tokens for linguistic content and fixed-length global tokens for speaker attributes. This disentangled representation, combined with the Qwen2.5 LLM and a chain-of-thought (CoT) generation approach, enables both coarse-grained control (e.g., gender, speaking style) and fine-grained adjustments (e.g., precise pitch values, speaking rate). To facilitate research in controllable TTS, we introduce VoxBox, a meticulously curated 100,000-hour dataset with comprehensive attribute annotations. Extensive experiments demonstrate that Spark-TTS not only achieves state-of-the-art zero-shot voice cloning but also generates highly customizable voices that surpass the limitations of reference-based synthesis. Source code, pre-trained models, and audio samples are available at https://github.com/SparkAudio/Spark-TTS.
Delta Decompression for MoE-based LLMs Compression
Feb 24, 2025



Mixture-of-Experts (MoE) architectures in large language models (LLMs) achieve exceptional performance, but face prohibitive storage and memory requirements. To address these challenges, we present $D^2$-MoE, a new delta decompression compressor for reducing the parameters of MoE LLMs. Based on observations of expert diversity, we decompose their weights into a shared base weight and unique delta weights. Specifically, our method first merges each expert's weight into the base weight using the Fisher information matrix to capture shared components. Then, we compress delta weights through Singular Value Decomposition (SVD) by exploiting their low-rank properties. Finally, we introduce a semi-dynamical structured pruning strategy for the base weights, combining static and dynamic redundancy analysis to achieve further parameter reduction while maintaining input adaptivity. In this way, our $D^2$-MoE successfully compact MoE LLMs to high compression ratios without additional training. Extensive experiments highlight the superiority of our approach, with over 13% performance gains than other compressors on Mixtral|Phi-3.5|DeepSeek|Qwen2 MoE LLMs at 40$\sim$60% compression rates. Codes are available in https://github.com/lliai/D2MoE.
Audio-FLAN: A Preliminary Release
Feb 23, 2025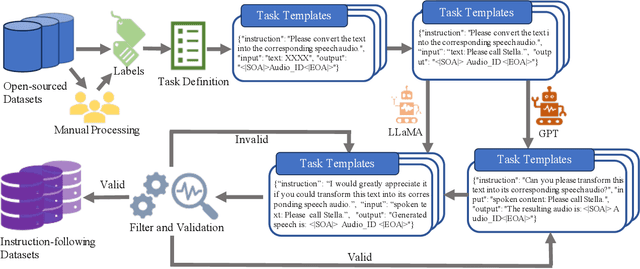

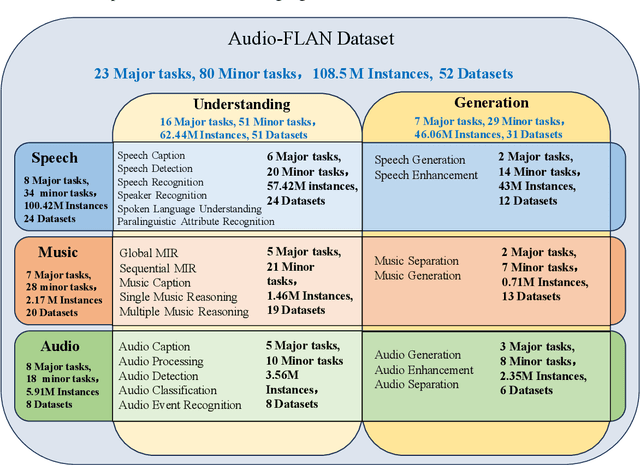

Recent advancements in audio tokenization have significantly enhanced the integration of audio capabilities into large language models (LLMs). However, audio understanding and generation are often treated as distinct tasks, hindering the development of truly unified audio-language models. While instruction tuning has demonstrated remarkable success in improving generalization and zero-shot learning across text and vision, its application to audio remains largely unexplored. A major obstacle is the lack of comprehensive datasets that unify audio understanding and generation. To address this, we introduce Audio-FLAN, a large-scale instruction-tuning dataset covering 80 diverse tasks across speech, music, and sound domains, with over 100 million instances. Audio-FLAN lays the foundation for unified audio-language models that can seamlessly handle both understanding (e.g., transcription, comprehension) and generation (e.g., speech, music, sound) tasks across a wide range of audio domains in a zero-shot manner. The Audio-FLAN dataset is available on HuggingFace and GitHub and will be continuously updated.
Machine learning for modelling unstructured grid data in computational physics: a review
Feb 13, 2025
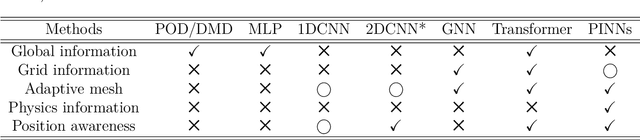
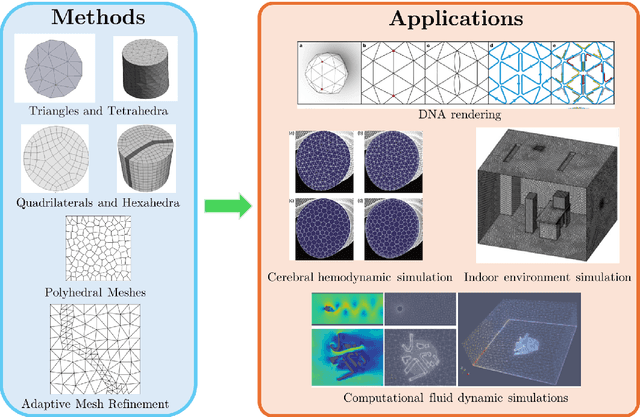

Unstructured grid data are essential for modelling complex geometries and dynamics in computational physics. Yet, their inherent irregularity presents significant challenges for conventional machine learning (ML) techniques. This paper provides a comprehensive review of advanced ML methodologies designed to handle unstructured grid data in high-dimensional dynamical systems. Key approaches discussed include graph neural networks, transformer models with spatial attention mechanisms, interpolation-integrated ML methods, and meshless techniques such as physics-informed neural networks. These methodologies have proven effective across diverse fields, including fluid dynamics and environmental simulations. This review is intended as a guidebook for computational scientists seeking to apply ML approaches to unstructured grid data in their domains, as well as for ML researchers looking to address challenges in computational physics. It places special focus on how ML methods can overcome the inherent limitations of traditional numerical techniques and, conversely, how insights from computational physics can inform ML development. To support benchmarking, this review also provides a summary of open-access datasets of unstructured grid data in computational physics. Finally, emerging directions such as generative models with unstructured data, reinforcement learning for mesh generation, and hybrid physics-data-driven paradigms are discussed to inspire future advancements in this evolving field.
VFX Creator: Animated Visual Effect Generation with Controllable Diffusion Transformer
Feb 09, 2025Crafting magic and illusions is one of the most thrilling aspects of filmmaking, with visual effects (VFX) serving as the powerhouse behind unforgettable cinematic experiences. While recent advances in generative artificial intelligence have driven progress in generic image and video synthesis, the domain of controllable VFX generation remains relatively underexplored. In this work, we propose a novel paradigm for animated VFX generation as image animation, where dynamic effects are generated from user-friendly textual descriptions and static reference images. Our work makes two primary contributions: (i) Open-VFX, the first high-quality VFX video dataset spanning 15 diverse effect categories, annotated with textual descriptions, instance segmentation masks for spatial conditioning, and start-end timestamps for temporal control. (ii) VFX Creator, a simple yet effective controllable VFX generation framework based on a Video Diffusion Transformer. The model incorporates a spatial and temporal controllable LoRA adapter, requiring minimal training videos. Specifically, a plug-and-play mask control module enables instance-level spatial manipulation, while tokenized start-end motion timestamps embedded in the diffusion process, alongside the text encoder, allow precise temporal control over effect timing and pace. Extensive experiments on the Open-VFX test set demonstrate the superiority of the proposed system in generating realistic and dynamic effects, achieving state-of-the-art performance and generalization ability in both spatial and temporal controllability. Furthermore, we introduce a specialized metric to evaluate the precision of temporal control. By bridging traditional VFX techniques with generative approaches, VFX Creator unlocks new possibilities for efficient and high-quality video effect generation, making advanced VFX accessible to a broader audience.