 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDe Novo Molecular Generation via Connection-aware Motif Mining
Feb 02, 2023De novo molecular generation is an essential task for science discovery. Recently, fragment-based deep generative models have attracted much research attention due to their flexibility in generating novel molecules based on existing molecule fragments. However, the motif vocabulary, i.e., the collection of frequent fragments, is usually built upon heuristic rules, which brings difficulties to capturing common substructures from large amounts of molecules. In this work, we propose a new method, MiCaM, to generate molecules based on mined connection-aware motifs. Specifically, it leverages a data-driven algorithm to automatically discover motifs from a molecule library by iteratively merging subgraphs based on their frequency. The obtained motif vocabulary consists of not only molecular motifs (i.e., the frequent fragments), but also their connection information, indicating how the motifs are connected with each other. Based on the mined connection-aware motifs, MiCaM builds a connection-aware generator, which simultaneously picks up motifs and determines how they are connected. We test our method on distribution-learning benchmarks (i.e., generating novel molecules to resemble the distribution of a given training set) and goal-directed benchmarks (i.e., generating molecules with target properties), and achieve significant improvements over previous fragment-based baselines. Furthermore, we demonstrate that our method can effectively mine domain-specific motifs for different tasks.
Retrosynthetic Planning with Dual Value Networks
Jan 31, 2023Retrosynthesis, which aims to find a route to synthesize a target molecule from commercially available starting materials, is a critical task in drug discovery and materials design. Recently, the combination of ML-based single-step reaction predictors with multi-step planners has led to promising results. However, the single-step predictors are mostly trained offline to optimize the single-step accuracy, without considering complete routes. Here, we leverage reinforcement learning (RL) to improve the single-step predictor, by using a tree-shaped MDP to optimize complete routes while retaining single-step accuracy. Desirable routes should be both synthesizable and of low cost. We propose an online training algorithm, called Planning with Dual Value Networks (PDVN), in which two value networks predict the synthesizability and cost of molecules, respectively. To maintain the single-step accuracy, we design a two-branch network structure for the single-step predictor. On the widely-used USPTO dataset, our PDVN algorithm improves the search success rate of existing multi-step planners (e.g., increasing the success rate from 85.79% to 98.95% for Retro*, and reducing the number of model calls by half while solving 99.47% molecules for RetroGraph). Furthermore, PDVN finds shorter synthesis routes (e.g., reducing the average route length from 5.76 to 4.83 for Retro*, and from 5.63 to 4.78 for RetroGraph).
Incorporating Pre-training Paradigm for Antibody Sequence-Structure Co-design
Nov 17, 2022Antibodies are versatile proteins that can bind to pathogens and provide effective protection for human body. Recently, deep learning-based computational antibody design has attracted popular attention since it automatically mines the antibody patterns from data that could be complementary to human experiences. However, the computational methods heavily rely on high-quality antibody structure data, which is quite limited. Besides, the complementarity-determining region (CDR), which is the key component of an antibody that determines the specificity and binding affinity, is highly variable and hard to predict. Therefore, the data limitation issue further raises the difficulty of CDR generation for antibodies. Fortunately, there exists a large amount of sequence data of antibodies that can help model the CDR and alleviate the reliance on structure data. By witnessing the success of pre-training models for protein modeling, in this paper, we develop the antibody pre-training language model and incorporate it into the (antigen-specific) antibody design model in a systemic way. Specifically, we first pre-train an antibody language model based on the sequence data, then propose a one-shot way for sequence and structure generation of CDR to avoid the heavy cost and error propagation from an autoregressive manner, and finally leverage the pre-trained antibody model for the antigen-specific antibody generation model with some carefully designed modules. Through various experiments, we show that our method achieves superior performances over previous baselines on different tasks, such as sequence and structure generation and antigen-binding CDR-H3 design.
BioGPT: Generative Pre-trained Transformer for Biomedical Text Generation and Mining
Oct 19, 2022
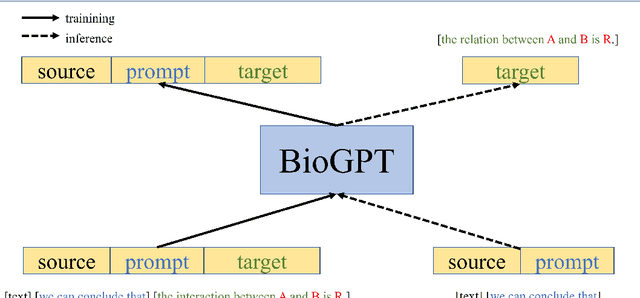
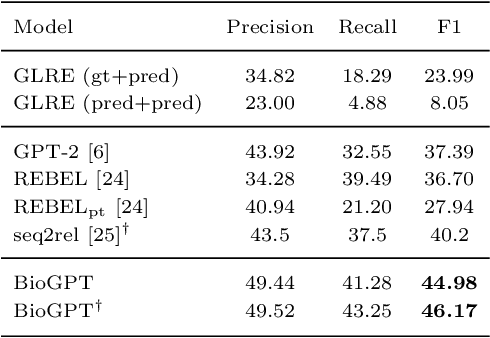
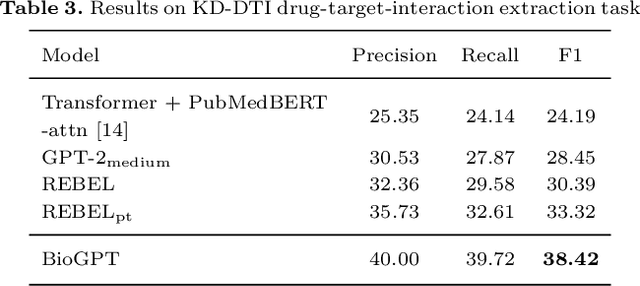
Pre-trained language models have attracted increasing attention in the biomedical domain, inspired by their great success in the general natural language domain. Among the two main branches of pre-trained language models in the general language domain, i.e., BERT (and its variants) and GPT (and its variants), the first one has been extensively studied in the biomedical domain, such as BioBERT and PubMedBERT. While they have achieved great success on a variety of discriminative downstream biomedical tasks, the lack of generation ability constrains their application scope. In this paper, we propose BioGPT, a domain-specific generative Transformer language model pre-trained on large scale biomedical literature. We evaluate BioGPT on six biomedical NLP tasks and demonstrate that our model outperforms previous models on most tasks. Especially, we get 44.98%, 38.42% and 40.76% F1 score on BC5CDR, KD-DTI and DDI end-to-end relation extraction tasks respectively, and 78.2% accuracy on PubMedQA, creating a new record. Our case study on text generation further demonstrates the advantage of BioGPT on biomedical literature to generate fluent descriptions for biomedical terms. Code is available at https://github.com/microsoft/BioGPT.
* Published at Briefings in Bioinformatics. Code is available at https://github.com/microsoft/BioGPT
Unified 2D and 3D Pre-Training of Molecular Representations
Jul 14, 2022
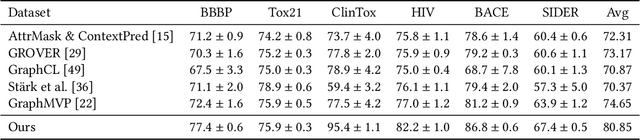
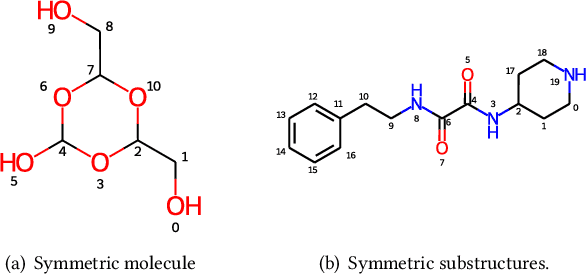
Molecular representation learning has attracted much attention recently. A molecule can be viewed as a 2D graph with nodes/atoms connected by edges/bonds, and can also be represented by a 3D conformation with 3-dimensional coordinates of all atoms. We note that most previous work handles 2D and 3D information separately, while jointly leveraging these two sources may foster a more informative representation. In this work, we explore this appealing idea and propose a new representation learning method based on a unified 2D and 3D pre-training. Atom coordinates and interatomic distances are encoded and then fused with atomic representations through graph neural networks. The model is pre-trained on three tasks: reconstruction of masked atoms and coordinates, 3D conformation generation conditioned on 2D graph, and 2D graph generation conditioned on 3D conformation. We evaluate our method on 11 downstream molecular property prediction tasks: 7 with 2D information only and 4 with both 2D and 3D information. Our method achieves state-of-the-art results on 10 tasks, and the average improvement on 2D-only tasks is 8.3%. Our method also achieves significant improvement on two 3D conformation generation tasks.
RetroGraph: Retrosynthetic Planning with Graph Search
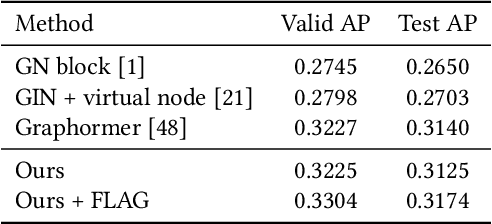
Jun 23, 2022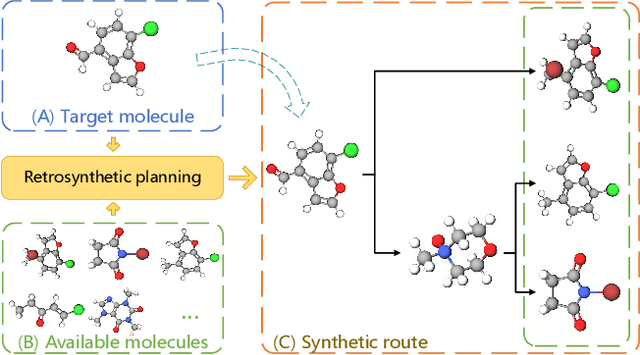
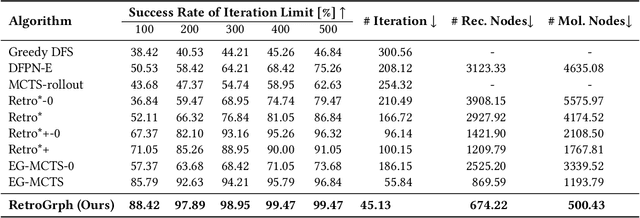
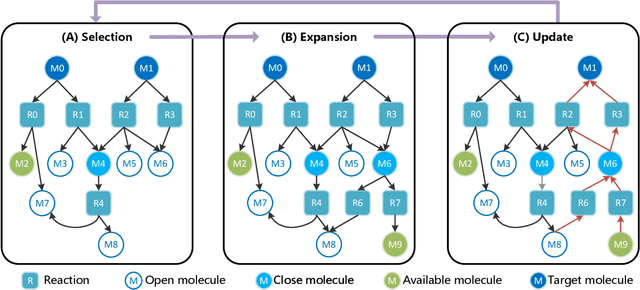
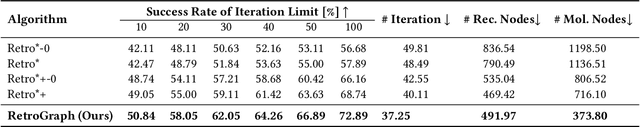
Retrosynthetic planning, which aims to find a reaction pathway to synthesize a target molecule, plays an important role in chemistry and drug discovery. This task is usually modeled as a search problem. Recently, data-driven methods have attracted many research interests and shown promising results for retrosynthetic planning. We observe that the same intermediate molecules are visited many times in the searching process, and they are usually independently treated in previous tree-based methods (e.g., AND-OR tree search, Monte Carlo tree search). Such redundancies make the search process inefficient. We propose a graph-based search policy that eliminates the redundant explorations of any intermediate molecules. As searching over a graph is more complicated than over a tree, we further adopt a graph neural network to guide the search over graphs. Meanwhile, our method can search a batch of targets together in the graph and remove the inter-target duplication in the tree-based search methods. Experimental results on two datasets demonstrate the effectiveness of our method. Especially on the widely used USPTO benchmark, we improve the search success rate to 99.47%, advancing previous state-of-the-art performance for 2.6 points.
SMT-DTA: Improving Drug-Target Affinity Prediction with Semi-supervised Multi-task Training
Jun 22, 2022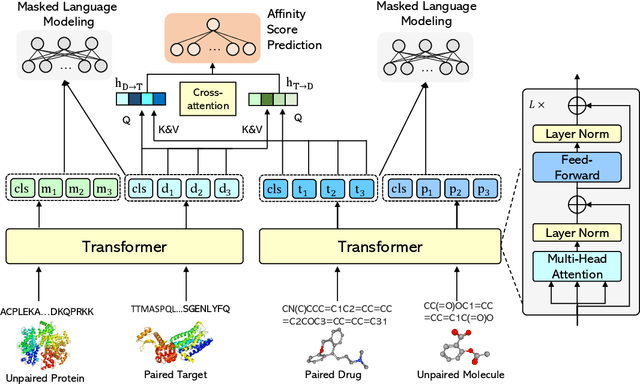
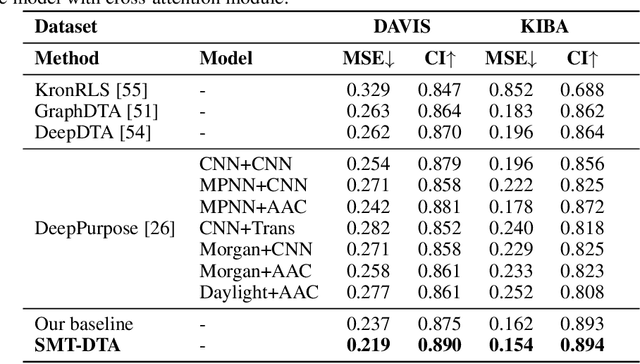
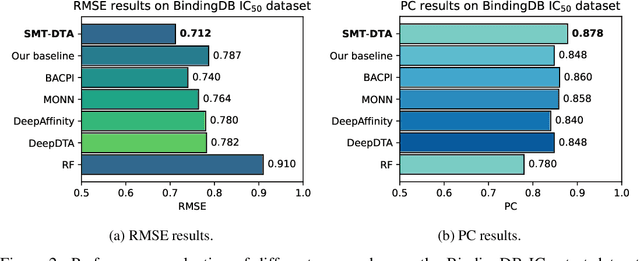

Drug-Target Affinity (DTA) prediction is an essential task for drug discovery and pharmaceutical research. Accurate predictions of DTA can greatly benefit the design of new drug. As wet experiments are costly and time consuming, the supervised data for DTA prediction is extremely limited. This seriously hinders the application of deep learning based methods, which require a large scale of supervised data. To address this challenge and improve the DTA prediction accuracy, we propose a framework with several simple yet effective strategies in this work: (1) a multi-task training strategy, which takes the DTA prediction and the masked language modeling (MLM) task on the paired drug-target dataset; (2) a semi-supervised training method to empower the drug and target representation learning by leveraging large-scale unpaired molecules and proteins in training, which differs from previous pre-training and fine-tuning methods that only utilize molecules or proteins in pre-training; and (3) a cross-attention module to enhance the interaction between drug and target representation. Extensive experiments are conducted on three real-world benchmark datasets: BindingDB, DAVIS and KIBA. The results show that our framework significantly outperforms existing methods and achieves state-of-the-art performances, e.g., $0.712$ RMSE on BindingDB IC$_{50}$ measurement with more than $5\%$ improvement than previous best work. In addition, case studies on specific drug-target binding activities, drug feature visualizations, and real-world applications demonstrate the great potential of our work. The code and data are released at https://github.com/QizhiPei/SMT-DTA
Direct Molecular Conformation Generation
Feb 03, 2022
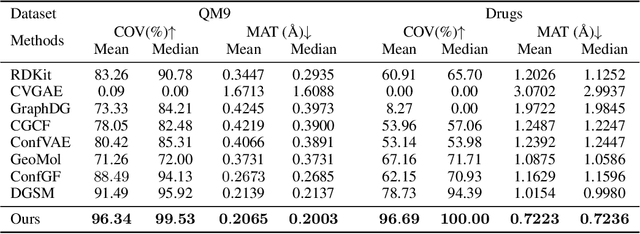
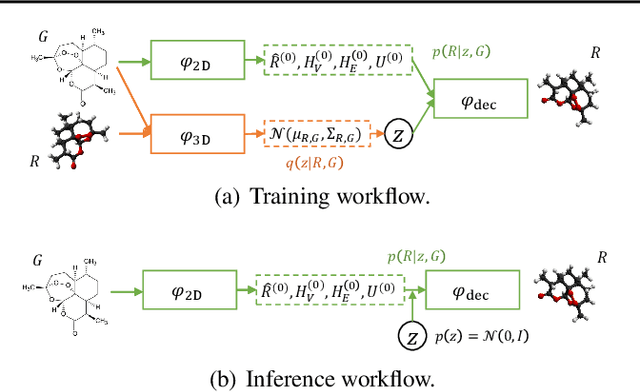
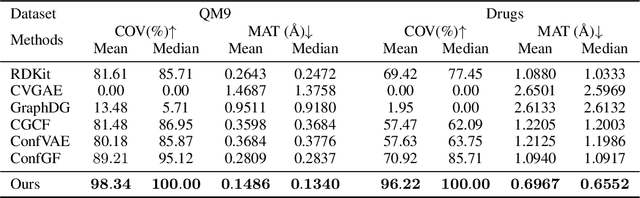
Molecular conformation generation aims to generate three-dimensional coordinates of all the atoms in a molecule and is an important task in bioinformatics and pharmacology. Previous distance-based methods first predict interatomic distances and then generate conformations based on them, which could result in conflicting distances. In this work, we propose a method that directly predicts the coordinates of atoms. We design a dedicated loss function for conformation generation, which is invariant to roto-translation of coordinates of conformations and permutation of symmetric atoms in molecules. We further design a backbone model that stacks multiple blocks, where each block refines the conformation generated by its preceding block. Our method achieves state-of-the-art results on four public benchmarks: on small-scale GEOM-QM9 and GEOM-Drugs which have $200$K training data, we can improve the previous best matching score by $3.5\%$ and $28.9\%$; on large-scale GEOM-QM9 and GEOM-Drugs which have millions of training data, those two improvements are $47.1\%$ and $36.3\%$. This shows the effectiveness of our method and the great potential of the direct approach. Our code is released at \url{https://github.com/DirectMolecularConfGen/DMCG}.
DDG-DA: Data Distribution Generation for Predictable Concept Drift Adaptation
Jan 11, 2022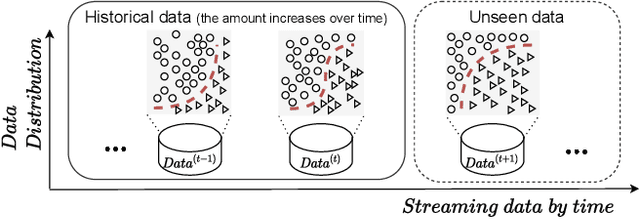
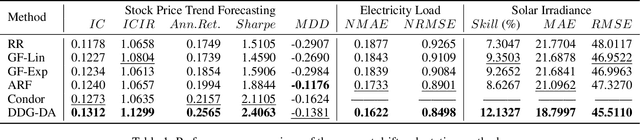
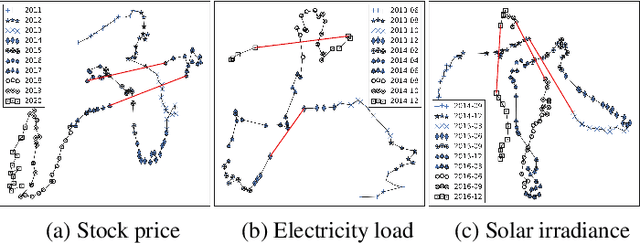
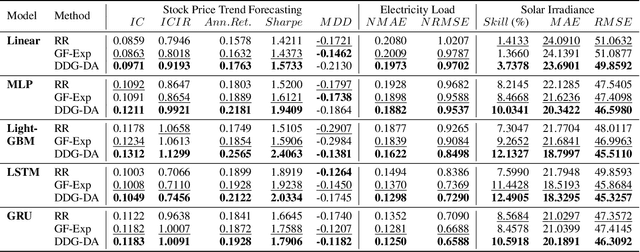
In many real-world scenarios, we often deal with streaming data that is sequentially collected over time. Due to the non-stationary nature of the environment, the streaming data distribution may change in unpredictable ways, which is known as concept drift. To handle concept drift, previous methods first detect when/where the concept drift happens and then adapt models to fit the distribution of the latest data. However, there are still many cases that some underlying factors of environment evolution are predictable, making it possible to model the future concept drift trend of the streaming data, while such cases are not fully explored in previous work. In this paper, we propose a novel method DDG-DA, that can effectively forecast the evolution of data distribution and improve the performance of models. Specifically, we first train a predictor to estimate the future data distribution, then leverage it to generate training samples, and finally train models on the generated data. We conduct experiments on three real-world tasks (forecasting on stock price trend, electricity load and solar irradiance) and obtain significant improvement on multiple widely-used models.
SHGNN: Structure-Aware Heterogeneous Graph Neural Network
Dec 14, 2021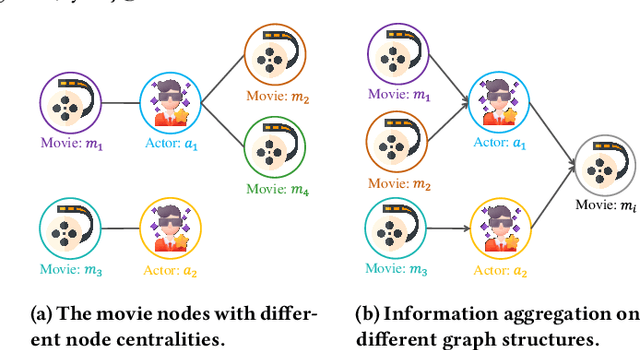
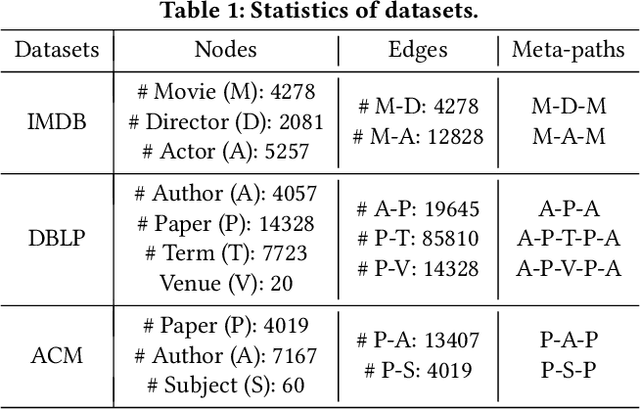
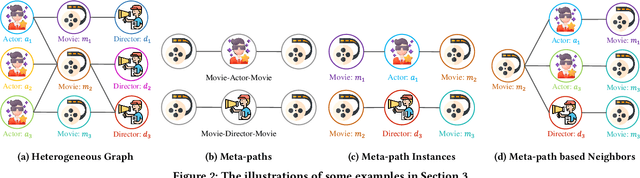
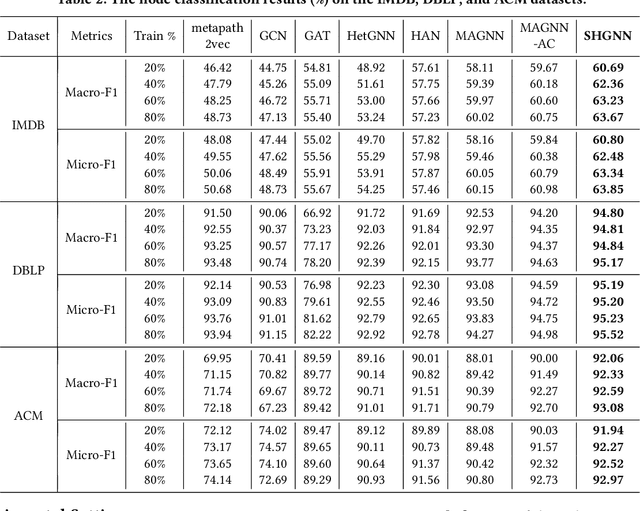
Many real-world graphs (networks) are heterogeneous with different types of nodes and edges. Heterogeneous graph embedding, aiming at learning the low-dimensional node representations of a heterogeneous graph, is vital for various downstream applications. Many meta-path based embedding methods have been proposed to learn the semantic information of heterogeneous graphs in recent years. However, most of the existing techniques overlook the graph structure information when learning the heterogeneous graph embeddings. This paper proposes a novel Structure-Aware Heterogeneous Graph Neural Network (SHGNN) to address the above limitations. In detail, we first utilize a feature propagation module to capture the local structure information of intermediate nodes in the meta-path. Next, we use a tree-attention aggregator to incorporate the graph structure information into the aggregation module on the meta-path. Finally, we leverage a meta-path aggregator to fuse the information aggregated from different meta-paths. We conducted experiments on node classification and clustering tasks and achieved state-of-the-art results on the benchmark datasets, which shows the effectiveness of our proposed method.