 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTrustLLM: Trustworthiness in Large Language Models
Jan 25, 2024



Large language models (LLMs), exemplified by ChatGPT, have gained considerable attention for their excellent natural language processing capabilities. Nonetheless, these LLMs present many challenges, particularly in the realm of trustworthiness. Therefore, ensuring the trustworthiness of LLMs emerges as an important topic. This paper introduces TrustLLM, a comprehensive study of trustworthiness in LLMs, including principles for different dimensions of trustworthiness, established benchmark, evaluation, and analysis of trustworthiness for mainstream LLMs, and discussion of open challenges and future directions. Specifically, we first propose a set of principles for trustworthy LLMs that span eight different dimensions. Based on these principles, we further establish a benchmark across six dimensions including truthfulness, safety, fairness, robustness, privacy, and machine ethics. We then present a study evaluating 16 mainstream LLMs in TrustLLM, consisting of over 30 datasets. Our findings firstly show that in general trustworthiness and utility (i.e., functional effectiveness) are positively related. Secondly, our observations reveal that proprietary LLMs generally outperform most open-source counterparts in terms of trustworthiness, raising concerns about the potential risks of widely accessible open-source LLMs. However, a few open-source LLMs come very close to proprietary ones. Thirdly, it is important to note that some LLMs may be overly calibrated towards exhibiting trustworthiness, to the extent that they compromise their utility by mistakenly treating benign prompts as harmful and consequently not responding. Finally, we emphasize the importance of ensuring transparency not only in the models themselves but also in the technologies that underpin trustworthiness. Knowing the specific trustworthy technologies that have been employed is crucial for analyzing their effectiveness.
Let's Think Outside the Box: Exploring Leap-of-Thought in Large Language Models with Creative Humor Generation
Dec 06, 2023



Chain-of-Thought (CoT) guides large language models (LLMs) to reason step-by-step, and can motivate their logical reasoning ability. While effective for logical tasks, CoT is not conducive to creative problem-solving which often requires out-of-box thoughts and is crucial for innovation advancements. In this paper, we explore the Leap-of-Thought (LoT) abilities within LLMs -- a non-sequential, creative paradigm involving strong associations and knowledge leaps. To this end, we study LLMs on the popular Oogiri game which needs participants to have good creativity and strong associative thinking for responding unexpectedly and humorously to the given image, text, or both, and thus is suitable for LoT study. Then to investigate LLMs' LoT ability in the Oogiri game, we first build a multimodal and multilingual Oogiri-GO dataset which contains over 130,000 samples from the Oogiri game, and observe the insufficient LoT ability or failures of most existing LLMs on the Oogiri game. Accordingly, we introduce a creative Leap-of-Thought (CLoT) paradigm to improve LLM's LoT ability. CLoT first formulates the Oogiri-GO dataset into LoT-oriented instruction tuning data to train pretrained LLM for achieving certain LoT humor generation and discrimination abilities. Then CLoT designs an explorative self-refinement that encourages the LLM to generate more creative LoT data via exploring parallels between seemingly unrelated concepts and selects high-quality data to train itself for self-refinement. CLoT not only excels in humor generation in the Oogiri game but also boosts creative abilities in various tasks like cloud guessing game and divergent association task. These findings advance our understanding and offer a pathway to improve LLMs' creative capacities for innovative applications across domains. The dataset, code, and models will be released online. https://zhongshsh.github.io/CLoT/.
Graph AI in Medicine
Oct 20, 2023In clinical artificial intelligence (AI), graph representation learning, mainly through graph neural networks (GNNs), stands out for its capability to capture intricate relationships within structured clinical datasets. With diverse data -- from patient records to imaging -- GNNs process data holistically by viewing modalities as nodes interconnected by their relationships. Graph AI facilitates model transfer across clinical tasks, enabling models to generalize across patient populations without additional parameters or minimal re-training. However, the importance of human-centered design and model interpretability in clinical decision-making cannot be overstated. Since graph AI models capture information through localized neural transformations defined on graph relationships, they offer both an opportunity and a challenge in elucidating model rationale. Knowledge graphs can enhance interpretability by aligning model-driven insights with medical knowledge. Emerging graph models integrate diverse data modalities through pre-training, facilitate interactive feedback loops, and foster human-AI collaboration, paving the way to clinically meaningful predictions.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023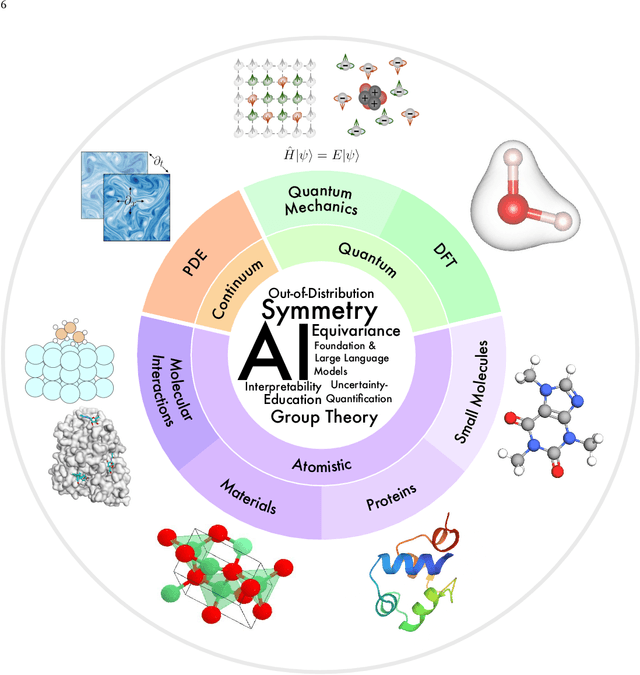



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Encoding Time-Series Explanations through Self-Supervised Model Behavior Consistency
Jun 03, 2023Interpreting time series models is uniquely challenging because it requires identifying both the location of time series signals that drive model predictions and their matching to an interpretable temporal pattern. While explainers from other modalities can be applied to time series, their inductive biases do not transfer well to the inherently uninterpretable nature of time series. We present TimeX, a time series consistency model for training explainers. TimeX trains an interpretable surrogate to mimic the behavior of a pretrained time series model. It addresses the issue of model faithfulness by introducing model behavior consistency, a novel formulation that preserves relations in the latent space induced by the pretrained model with relations in the latent space induced by TimeX. TimeX provides discrete attribution maps and, unlike existing interpretability methods, it learns a latent space of explanations that can be used in various ways, such as to provide landmarks to visually aggregate similar explanations and easily recognize temporal patterns. We evaluate TimeX on 8 synthetic and real-world datasets and compare its performance against state-of-the-art interpretability methods. We also conduct case studies using physiological time series. Quantitative evaluations demonstrate that TimeX achieves the highest or second-highest performance in every metric compared to baselines across all datasets. Through case studies, we show that the novel components of TimeX show potential for training faithful, interpretable models that capture the behavior of pretrained time series models.
GNNDelete: A General Strategy for Unlearning in Graph Neural Networks
Feb 26, 2023



Graph unlearning, which involves deleting graph elements such as nodes, node labels, and relationships from a trained graph neural network (GNN) model, is crucial for real-world applications where data elements may become irrelevant, inaccurate, or privacy-sensitive. However, existing methods for graph unlearning either deteriorate model weights shared across all nodes or fail to effectively delete edges due to their strong dependence on local graph neighborhoods. To address these limitations, we introduce GNNDelete, a novel model-agnostic layer-wise operator that optimizes two critical properties, namely, Deleted Edge Consistency and Neighborhood Influence, for graph unlearning. Deleted Edge Consistency ensures that the influence of deleted elements is removed from both model weights and neighboring representations, while Neighborhood Influence guarantees that the remaining model knowledge is preserved after deletion. GNNDelete updates representations to delete nodes and edges from the model while retaining the rest of the learned knowledge. We conduct experiments on seven real-world graphs, showing that GNNDelete outperforms existing approaches by up to 38.8% (AUC) on edge, node, and node feature deletion tasks, and 32.2% on distinguishing deleted edges from non-deleted ones. Additionally, GNNDelete is efficient, taking 12.3x less time and 9.3x less space than retraining GNN from scratch on WordNet18.
Domain Adaptation for Time Series Under Feature and Label Shifts
Feb 06, 2023



The transfer of models trained on labeled datasets in a source domain to unlabeled target domains is made possible by unsupervised domain adaptation (UDA). However, when dealing with complex time series models, the transferability becomes challenging due to the dynamic temporal structure that varies between domains, resulting in feature shifts and gaps in the time and frequency representations. Furthermore, tasks in the source and target domains can have vastly different label distributions, making it difficult for UDA to mitigate label shifts and recognize labels that only exist in the target domain. We present RAINCOAT, the first model for both closed-set and universal DA on complex time series. RAINCOAT addresses feature and label shifts by considering both temporal and frequency features, aligning them across domains, and correcting for misalignments to facilitate the detection of private labels. Additionally,RAINCOAT improves transferability by identifying label shifts in target domains. Our experiments with 5 datasets and 13 state-of-the-art UDA methods demonstrate that RAINCOAT can achieve an improvement in performance of up to 16.33%, and can effectively handle both closed-set and universal adaptation.
Geometric multimodal representation learning
Sep 07, 2022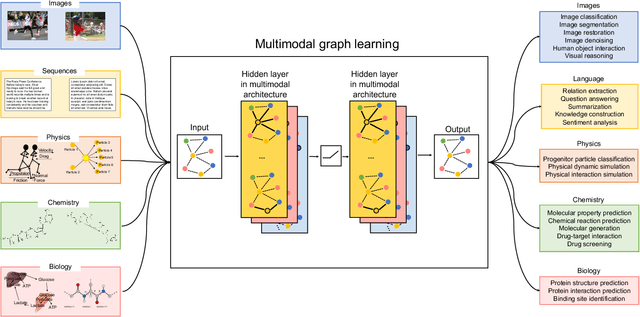
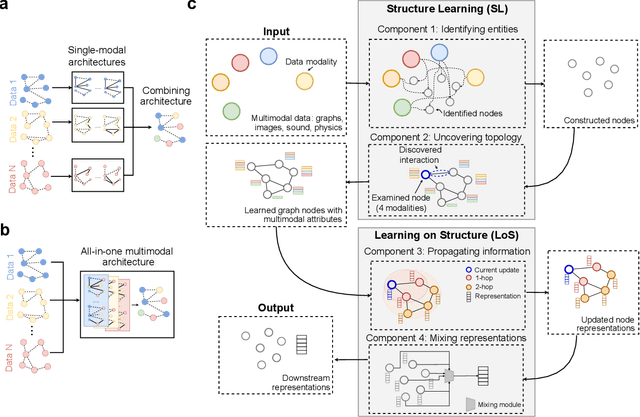
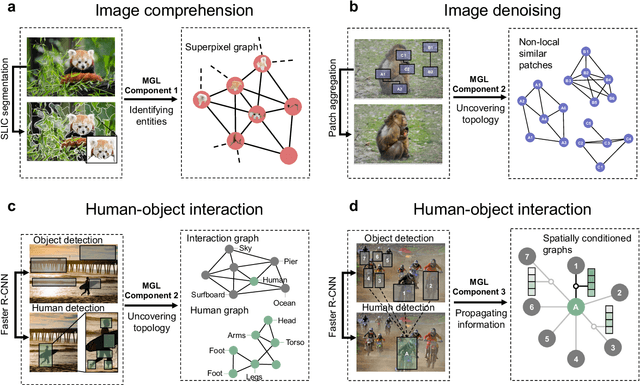
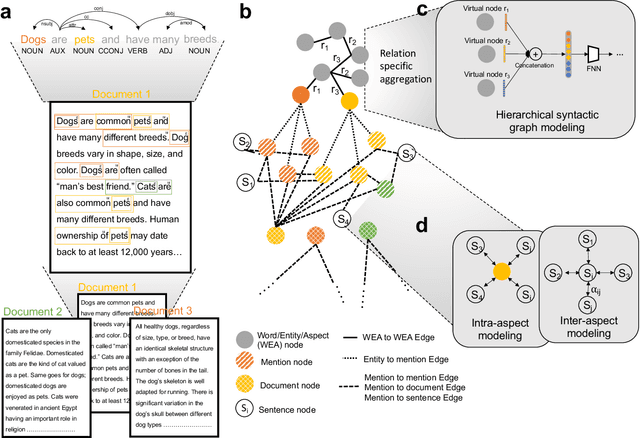
Graph-centric artificial intelligence (graph AI) has achieved remarkable success in modeling interacting systems prevalent in nature, from dynamical systems in biology to particle physics. The increasing heterogeneity of data calls for graph neural architectures that can combine multiple inductive biases. However, combining data from various sources is challenging because appropriate inductive bias may vary by data modality. Multimodal learning methods fuse multiple data modalities while leveraging cross-modal dependencies to address this challenge. Here, we survey 140 studies in graph-centric AI and realize that diverse data types are increasingly brought together using graphs and fed into sophisticated multimodal models. These models stratify into image-, language-, and knowledge-grounded multimodal learning. We put forward an algorithmic blueprint for multimodal graph learning based on this categorization. The blueprint serves as a way to group state-of-the-art architectures that treat multimodal data by choosing appropriately four different components. This effort can pave the way for standardizing the design of sophisticated multimodal architectures for highly complex real-world problems.
Evaluating Explainability for Graph Neural Networks
Aug 19, 2022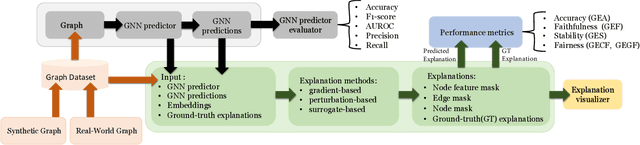
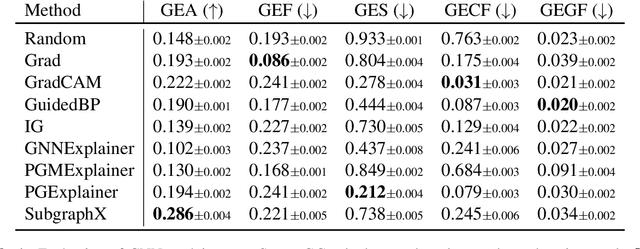
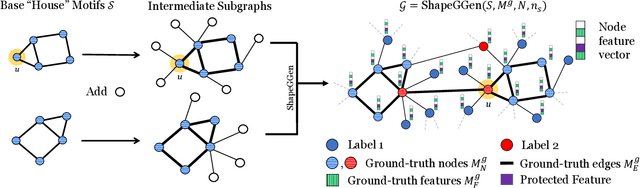

As post hoc explanations are increasingly used to understand the behavior of graph neural networks (GNNs), it becomes crucial to evaluate the quality and reliability of GNN explanations. However, assessing the quality of GNN explanations is challenging as existing graph datasets have no or unreliable ground-truth explanations for a given task. Here, we introduce a synthetic graph data generator, ShapeGGen, which can generate a variety of benchmark datasets (e.g., varying graph sizes, degree distributions, homophilic vs. heterophilic graphs) accompanied by ground-truth explanations. Further, the flexibility to generate diverse synthetic datasets and corresponding ground-truth explanations allows us to mimic the data generated by various real-world applications. We include ShapeGGen and several real-world graph datasets into an open-source graph explainability library, GraphXAI. In addition to synthetic and real-world graph datasets with ground-truth explanations, GraphXAI provides data loaders, data processing functions, visualizers, GNN model implementations, and evaluation metrics to benchmark the performance of GNN explainability methods.
OpenXAI: Towards a Transparent Evaluation of Model Explanations
Jun 22, 2022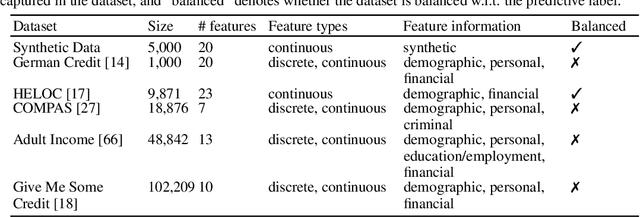
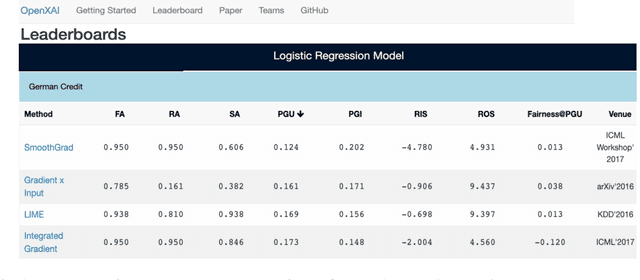
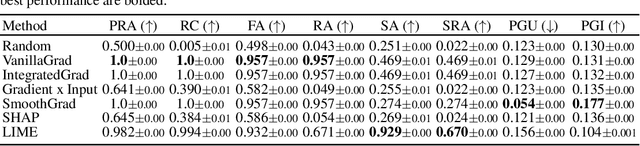
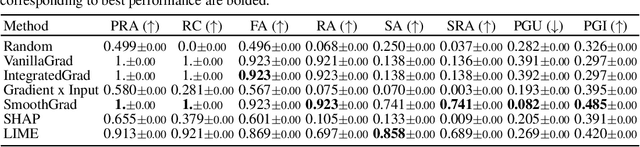
While several types of post hoc explanation methods (e.g., feature attribution methods) have been proposed in recent literature, there is little to no work on systematically benchmarking these methods in an efficient and transparent manner. Here, we introduce OpenXAI, a comprehensive and extensible open source framework for evaluating and benchmarking post hoc explanation methods. OpenXAI comprises of the following key components: (i) a flexible synthetic data generator and a collection of diverse real-world datasets, pre-trained models, and state-of-the-art feature attribution methods, (ii) open-source implementations of twenty-two quantitative metrics for evaluating faithfulness, stability (robustness), and fairness of explanation methods, and (iii) the first ever public XAI leaderboards to benchmark explanations. OpenXAI is easily extensible, as users can readily evaluate custom explanation methods and incorporate them into our leaderboards. Overall, OpenXAI provides an automated end-to-end pipeline that not only simplifies and standardizes the evaluation of post hoc explanation methods, but also promotes transparency and reproducibility in benchmarking these methods. OpenXAI datasets and data loaders, implementations of state-of-the-art explanation methods and evaluation metrics, as well as leaderboards are publicly available at https://open-xai.github.io/.