 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhylaFlow: Hybrid Flow Matching in Billera-Holmes-Vogtmann Tree Space for Phylogenetic Inference
May 21, 2026Phylogenetic trees are hybrid objects: branch lengths vary continuously, while topologies change discretely through edge contractions and expansions. Billera-Holmes-Vogtmann (BHV) tree space provides a canonical geometry for this structure, representing each resolved topology as a Euclidean orthant and topological changes as motion across shared lower-dimensional boundaries. We introduce PhylaFlow, a hybrid flow-matching model that learns posterior-basin transport in BHV tree space. PhylaFlow is trained on BHV geodesic paths from random starting trees to short-run posterior samples, coupling continuous branch-length motion within orthants with learned boundary events and discrete topology transitions. We evaluate the learned geometry operationally: if the flow reaches posterior-relevant regions, finite-budget Bayesian refinement initialized from, or guided by, its terminal trees should recover posterior-supported topologies more efficiently. Across DS1-DS8 phylogenetic posterior benchmarks, PhylaFlow substantially reduces initial Tree-KL relative to classical initializers. After finite-budget MrBayes refinement, direct PhylaFlow improves early and intermediate topology-recovery trajectories on most datasets, while split-guided PhylaFlow-MCMC obtains the strongest hard-case results. The best PhylaFlow variant outperforms short-warmup on seven of eight datasets and PhyloGFN on five of eight under the same refinement budget. In a joint sequence-conditioned experiment, sequence embeddings steer posterior split recovery, although exact posterior topology recovery remains preliminary. These results show that hybrid flow matching can learn actionable transport in BHV tree space and provide a geometry-aware proposal mechanism for Bayesian phylogenetic inference.
Evaluating Relational Reasoning in LLMs with REL
Apr 14, 2026Relational reasoning is the ability to infer relations that jointly bind multiple entities, attributes, or variables. This ability is central to scientific reasoning, but existing evaluations of relational reasoning in large language models often focus on structured inputs such as tables, graphs, or synthetic tasks, and do not isolate the difficulty introduced by higher-arity relational binding. We study this problem through the lens of Relational Complexity (RC), which we define as the minimum number of independent entities or operands that must be simultaneously bound to apply a relation. RC provides a principled way to vary reasoning difficulty while controlling for confounders such as input size, vocabulary, and representational choices. Building on RC, we introduce REL, a generative benchmark framework spanning algebra, chemistry, and biology that varies RC within each domain. Across frontier LLMs, performance degrades consistently and monotonically as RC increases, even when the total number of entities is held fixed. This failure mode persists with increased test-time compute and in-context learning, suggesting a limitation tied to the arity of the required relational binding rather than to insufficient inference steps or lack of exposure to examples. Our results identify a regime of higher-arity reasoning in which current models struggle, and motivate re-examining benchmarks through the lens of relational complexity.
Controllable Sequence Editing for Counterfactual Generation
Feb 05, 2025



Sequence models generate counterfactuals by modifying parts of a sequence based on a given condition, enabling reasoning about "what if" scenarios. While these models excel at conditional generation, they lack fine-grained control over when and where edits occur. Existing approaches either focus on univariate sequences or assume that interventions affect the entire sequence globally. However, many applications require precise, localized modifications, where interventions take effect only after a specified time and impact only a subset of co-occurring variables. We introduce CLEF, a controllable sequence editing model for counterfactual reasoning about both immediate and delayed effects. CLEF learns temporal concepts that encode how and when interventions should influence a sequence. With these concepts, CLEF selectively edits relevant time steps while preserving unaffected portions of the sequence. We evaluate CLEF on cellular and patient trajectory datasets, where gene regulation affects only certain genes at specific time steps, or medical interventions alter only a subset of lab measurements. CLEF improves immediate sequence editing by up to 36.01% in MAE compared to baselines. Unlike prior methods, CLEF enables one-step generation of counterfactual sequences at any future time step, outperforming baselines by up to 65.71% in MAE. A case study on patients with type 1 diabetes mellitus shows that CLEF identifies clinical interventions that shift patient trajectories toward healthier outcomes.
Reinforcement learning on structure-conditioned categorical diffusion for protein inverse folding
Oct 22, 2024



Protein inverse folding-that is, predicting an amino acid sequence that will fold into the desired 3D structure-is an important problem for structure-based protein design. Machine learning based methods for inverse folding typically use recovery of the original sequence as the optimization objective. However, inverse folding is a one-to-many problem where several sequences can fold to the same structure. Moreover, for many practical applications, it is often desirable to have multiple, diverse sequences that fold into the target structure since it allows for more candidate sequences for downstream optimizations. Here, we demonstrate that although recent inverse folding methods show increased sequence recovery, their "foldable diversity"-i.e. their ability to generate multiple non-similar sequences that fold into the structures consistent with the target-does not increase. To address this, we present RL-DIF, a categorical diffusion model for inverse folding that is pre-trained on sequence recovery and tuned via reinforcement learning on structural consistency. We find that RL-DIF achieves comparable sequence recovery and structural consistency to benchmark models but shows greater foldable diversity: experiments show RL-DIF can achieve an foldable diversity of 29% on CATH 4.2, compared to 23% from models trained on the same dataset. The PyTorch model weights and sampling code are available on GitHub.
Empowering Biomedical Discovery with AI Agents
Apr 03, 2024



We envision 'AI scientists' as systems capable of skeptical learning and reasoning that empower biomedical research through collaborative agents that integrate machine learning tools with experimental platforms. Rather than taking humans out of the discovery process, biomedical AI agents combine human creativity and expertise with AI's ability to analyze large datasets, navigate hypothesis spaces, and execute repetitive tasks. AI agents are proficient in a variety of tasks, including self-assessment and planning of discovery workflows. These agents use large language models and generative models to feature structured memory for continual learning and use machine learning tools to incorporate scientific knowledge, biological principles, and theories. AI agents can impact areas ranging from hybrid cell simulation, programmable control of phenotypes, and the design of cellular circuits to the development of new therapies.
Geometric multimodal representation learning
Sep 07, 2022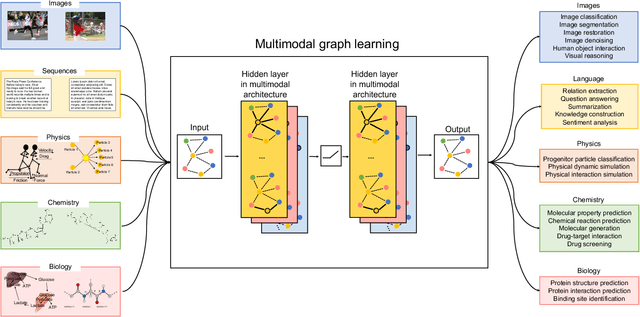
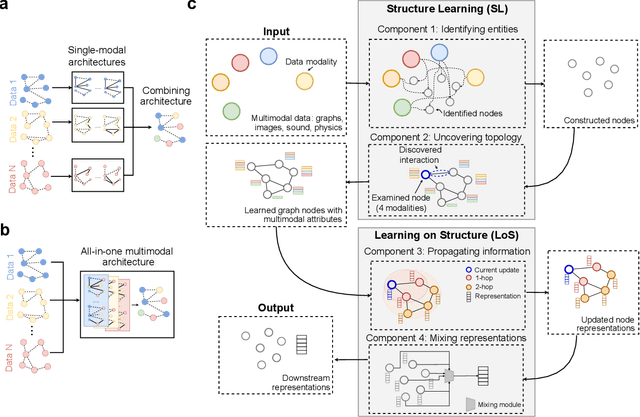
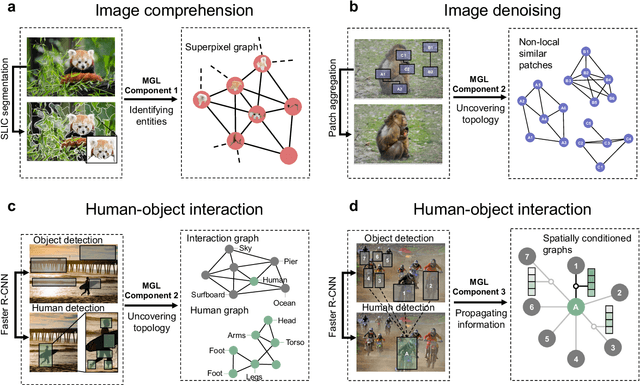
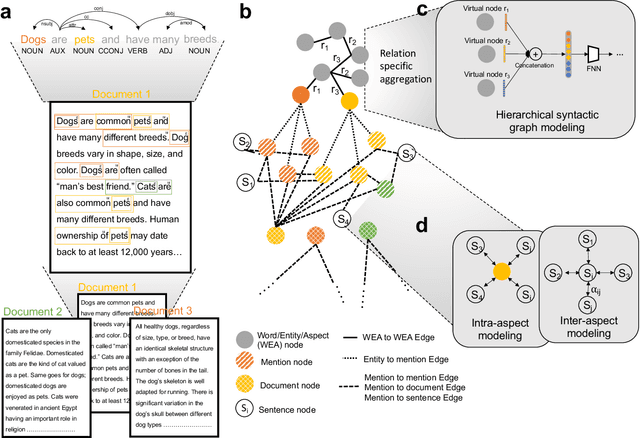
Graph-centric artificial intelligence (graph AI) has achieved remarkable success in modeling interacting systems prevalent in nature, from dynamical systems in biology to particle physics. The increasing heterogeneity of data calls for graph neural architectures that can combine multiple inductive biases. However, combining data from various sources is challenging because appropriate inductive bias may vary by data modality. Multimodal learning methods fuse multiple data modalities while leveraging cross-modal dependencies to address this challenge. Here, we survey 140 studies in graph-centric AI and realize that diverse data types are increasingly brought together using graphs and fed into sophisticated multimodal models. These models stratify into image-, language-, and knowledge-grounded multimodal learning. We put forward an algorithmic blueprint for multimodal graph learning based on this categorization. The blueprint serves as a way to group state-of-the-art architectures that treat multimodal data by choosing appropriately four different components. This effort can pave the way for standardizing the design of sophisticated multimodal architectures for highly complex real-world problems.