 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTIGER: Text-Informed Generalized Enzyme-Reaction Retrieval
May 23, 2026Enzyme-reaction retrieval is a fundamental problem in computational biology, underpinning enzyme characterization, reaction mechanism elucidation, and the rational design of metabolic pathways and biocatalysts. As a bidirectional task, it entails both enzyme-to-reaction and reaction-to-enzyme mapping. However, existing approaches suffer from poor generalization across tasks and distributions, with performance highly sensitive to dataset splits and substantial asymmetry between retrieval directions. To address these challenges, we present TIGER, a Text-Informed Generalized Enzyme-Reaction Retrieval framework that leverages protein-to-text generation models to distill textual semantic knowledge from enzyme sequences, providing a generalized representation that bridges enzymes and biochemical reactions. To ensure the quality and reliability of textual semantics, we design a Dynamic Gating Network that adaptively fuses text-derived knowledge with sequence features, enabling more consistent and informative enzyme representations, while a Structure-Shared Feature Projector aligns enzyme and reaction representations within a unified latent space. Extensive experiments demonstrate that, under bidirectional retrieval supervision, TIGER significantly outperforms state-of-the-art baselines across diverse distributions and exhibits strong robustness and transferability across tasks.
Embodied Science: Closing the Discovery Loop with Agentic Embodied AI
Mar 20, 2026Artificial intelligence has demonstrated remarkable capability in predicting scientific properties, yet scientific discovery remains an inherently physical, long-horizon pursuit governed by experimental cycles. Most current computational approaches are misaligned with this reality, framing discovery as isolated, task-specific predictions rather than continuous interaction with the physical world. Here, we argue for embodied science, a paradigm that reframes scientific discovery as a closed loop tightly coupling agentic reasoning with physical execution. We propose a unified Perception-Language-Action-Discovery (PLAD) framework, wherein embodied agents perceive experimental environments, reason over scientific knowledge, execute physical interventions, and internalize outcomes to drive subsequent exploration. By grounding computational reasoning in robust physical feedback, this approach bridges the gap between digital prediction and empirical validation, offering a roadmap for autonomous discovery systems in the life and chemical sciences.
InnoEval: On Research Idea Evaluation as a Knowledge-Grounded, Multi-Perspective Reasoning Problem
Feb 16, 2026The rapid evolution of Large Language Models has catalyzed a surge in scientific idea production, yet this leap has not been accompanied by a matching advance in idea evaluation. The fundamental nature of scientific evaluation needs knowledgeable grounding, collective deliberation, and multi-criteria decision-making. However, existing idea evaluation methods often suffer from narrow knowledge horizons, flattened evaluation dimensions, and the inherent bias in LLM-as-a-Judge. To address these, we regard idea evaluation as a knowledge-grounded, multi-perspective reasoning problem and introduce InnoEval, a deep innovation evaluation framework designed to emulate human-level idea assessment. We apply a heterogeneous deep knowledge search engine that retrieves and grounds dynamic evidence from diverse online sources. We further achieve review consensus with an innovation review board containing reviewers with distinct academic backgrounds, enabling a multi-dimensional decoupled evaluation across multiple metrics. We construct comprehensive datasets derived from authoritative peer-reviewed submissions to benchmark InnoEval. Experiments demonstrate that InnoEval can consistently outperform baselines in point-wise, pair-wise, and group-wise evaluation tasks, exhibiting judgment patterns and consensus highly aligned with human experts.
ClinDEF: A Dynamic Evaluation Framework for Large Language Models in Clinical Reasoning
Dec 29, 2025Clinical diagnosis begins with doctor-patient interaction, during which physicians iteratively gather information, determine examination and refine differential diagnosis through patients' response. This dynamic clinical-reasoning process is poorly represented by existing LLM benchmarks that focus on static question-answering. To mitigate these gaps, recent methods explore dynamic medical frameworks involving interactive clinical dialogues. Although effective, they often rely on limited, contamination-prone datasets and lack granular, multi-level evaluation. In this work, we propose ClinDEF, a dynamic framework for assessing clinical reasoning in LLMs through simulated diagnostic dialogues. Grounded in a disease knowledge graph, our method dynamically generates patient cases and facilitates multi-turn interactions between an LLM-based doctor and an automated patient agent. Our evaluation protocol goes beyond diagnostic accuracy by incorporating fine-grained efficiency analysis and rubric-based assessment of diagnostic quality. Experiments show that ClinDEF effectively exposes critical clinical reasoning gaps in state-of-the-art LLMs, offering a more nuanced and clinically meaningful evaluation paradigm.
Knowledge-Augmented Long-CoT Generation for Complex Biomolecular Reasoning
Nov 11, 2025



Understanding complex biomolecular mechanisms requires multi-step reasoning across molecular interactions, signaling cascades, and metabolic pathways. While large language models(LLMs) show promise in such tasks, their application to biomolecular problems is hindered by logical inconsistencies and the lack of grounding in domain knowledge. Existing approaches often exacerbate these issues: reasoning steps may deviate from biological facts or fail to capture long mechanistic dependencies. To address these challenges, we propose a Knowledge-Augmented Long-CoT Reasoning framework that integrates LLMs with knowledge graph-based multi-hop reasoning chains. The framework constructs mechanistic chains via guided multi-hop traversal and pruning on the knowledge graph; these chains are then incorporated into supervised fine-tuning to improve factual grounding and further refined with reinforcement learning to enhance reasoning reliability and consistency. Furthermore, to overcome the shortcomings of existing benchmarks, which are often restricted in scale and scope and lack annotations for deep reasoning chains, we introduce PrimeKGQA, a comprehensive benchmark for biomolecular question answering. Experimental results on both PrimeKGQA and existing datasets demonstrate that although larger closed-source models still perform well on relatively simple tasks, our method demonstrates clear advantages as reasoning depth increases, achieving state-of-the-art performance on multi-hop tasks that demand traversal of structured biological knowledge. These findings highlight the effectiveness of combining structured knowledge with advanced reasoning strategies for reliable and interpretable biomolecular reasoning.
Breaking the Modality Barrier: Generative Modeling for Accurate Molecule Retrieval from Mass Spectra
Nov 09, 2025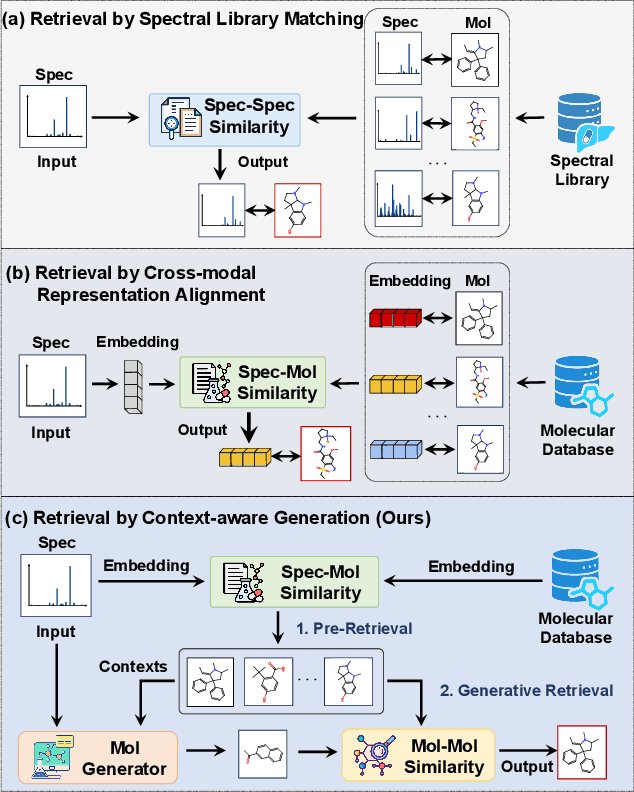



Retrieving molecular structures from tandem mass spectra is a crucial step in rapid compound identification. Existing retrieval methods, such as traditional mass spectral library matching, suffer from limited spectral library coverage, while recent cross-modal representation learning frameworks often encounter modality misalignment, resulting in suboptimal retrieval accuracy and generalization. To address these limitations, we propose GLMR, a Generative Language Model-based Retrieval framework that mitigates the cross-modal misalignment through a two-stage process. In the pre-retrieval stage, a contrastive learning-based model identifies top candidate molecules as contextual priors for the input mass spectrum. In the generative retrieval stage, these candidate molecules are integrated with the input mass spectrum to guide a generative model in producing refined molecular structures, which are then used to re-rank the candidates based on molecular similarity. Experiments on both MassSpecGym and the proposed MassRET-20k dataset demonstrate that GLMR significantly outperforms existing methods, achieving over 40% improvement in top-1 accuracy and exhibiting strong generalizability.
Copyright Protection for 3D Molecular Structures with Watermarking
Aug 25, 2025



Artificial intelligence (AI) revolutionizes molecule generation in bioengineering and biological research, significantly accelerating discovery processes. However, this advancement introduces critical concerns regarding intellectual property protection. To address these challenges, we propose the first robust watermarking method designed for molecules, which utilizes atom-level features to preserve molecular integrity and invariant features to ensure robustness against affine transformations. Comprehensive experiments validate the effectiveness of our method using the datasets QM9 and GEOM-DRUG, and generative models GeoBFN and GeoLDM. We demonstrate the feasibility of embedding watermarks, maintaining basic properties higher than 90.00\% while achieving watermark accuracy greater than 95.00\%. Furthermore, downstream docking simulations reveal comparable performance between original and watermarked molecules, with binding affinities reaching -6.00 kcal/mol and root mean square deviations below 1.602 \AA. These results confirm that our watermarking technique effectively safeguards molecular intellectual property without compromising scientific utility, enabling secure and responsible AI integration in molecular discovery and research applications.
SciToolAgent: A Knowledge Graph-Driven Scientific Agent for Multi-Tool Integration
Jul 27, 2025Scientific research increasingly relies on specialized computational tools, yet effectively utilizing these tools demands substantial domain expertise. While Large Language Models (LLMs) show promise in tool automation, they struggle to seamlessly integrate and orchestrate multiple tools for complex scientific workflows. Here, we present SciToolAgent, an LLM-powered agent that automates hundreds of scientific tools across biology, chemistry, and materials science. At its core, SciToolAgent leverages a scientific tool knowledge graph that enables intelligent tool selection and execution through graph-based retrieval-augmented generation. The agent also incorporates a comprehensive safety-checking module to ensure responsible and ethical tool usage. Extensive evaluations on a curated benchmark demonstrate that SciToolAgent significantly outperforms existing approaches. Case studies in protein engineering, chemical reactivity prediction, chemical synthesis, and metal-organic framework screening further demonstrate SciToolAgent's capability to automate complex scientific workflows, making advanced research tools accessible to both experts and non-experts.
KEPLA: A Knowledge-Enhanced Deep Learning Framework for Accurate Protein-Ligand Binding Affinity Prediction
Jun 16, 2025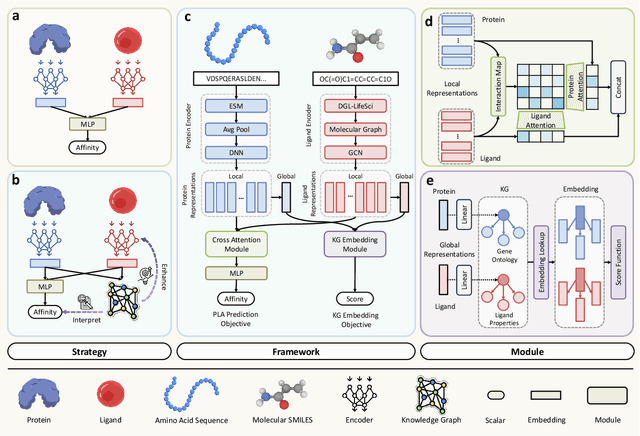
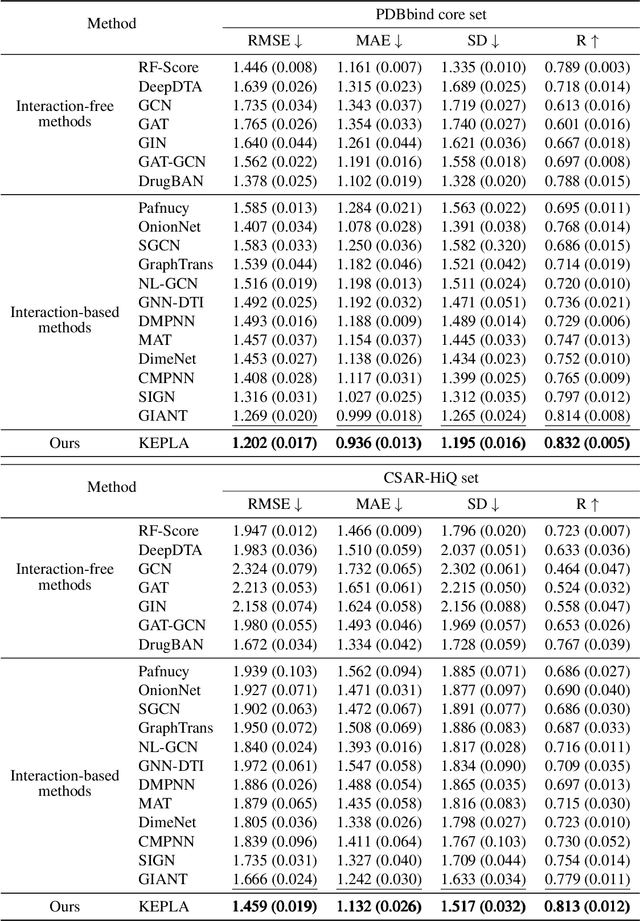
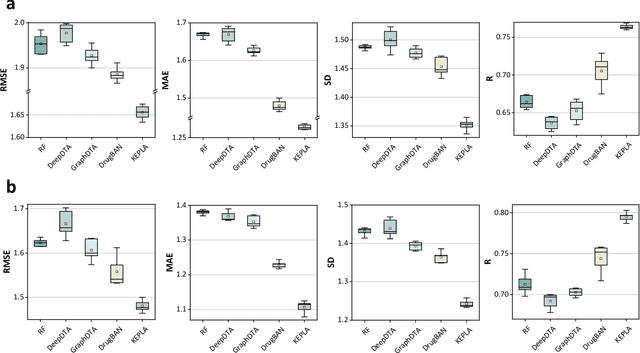
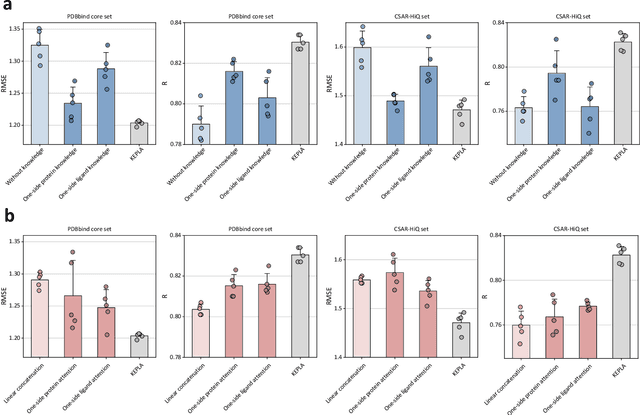
Accurate prediction of protein-ligand binding affinity is critical for drug discovery. While recent deep learning approaches have demonstrated promising results, they often rely solely on structural features, overlooking valuable biochemical knowledge associated with binding affinity. To address this limitation, we propose KEPLA, a novel deep learning framework that explicitly integrates prior knowledge from Gene Ontology and ligand properties of proteins and ligands to enhance prediction performance. KEPLA takes protein sequences and ligand molecular graphs as input and optimizes two complementary objectives: (1) aligning global representations with knowledge graph relations to capture domain-specific biochemical insights, and (2) leveraging cross attention between local representations to construct fine-grained joint embeddings for prediction. Experiments on two benchmark datasets across both in-domain and cross-domain scenarios demonstrate that KEPLA consistently outperforms state-of-the-art baselines. Furthermore, interpretability analyses based on knowledge graph relations and cross attention maps provide valuable insights into the underlying predictive mechanisms.
OneEval: Benchmarking LLM Knowledge-intensive Reasoning over Diverse Knowledge Bases
Jun 14, 2025



Large Language Models (LLMs) have demonstrated substantial progress on reasoning tasks involving unstructured text, yet their capabilities significantly deteriorate when reasoning requires integrating structured external knowledge such as knowledge graphs, code snippets, or formal logic. This limitation is partly due to the absence of benchmarks capable of systematically evaluating LLM performance across diverse structured knowledge modalities. To address this gap, we introduce \textbf{\textsc{OneEval}}, a comprehensive benchmark explicitly designed to assess the knowledge-intensive reasoning capabilities of LLMs across four structured knowledge modalities, unstructured text, knowledge graphs, code, and formal logic, and five critical domains (general knowledge, government, science, law, and programming). \textsc{OneEval} comprises 4,019 carefully curated instances and includes a challenging subset, \textsc{OneEval}\textsubscript{Hard}, consisting of 1,285 particularly difficult cases. Through extensive evaluation of 18 state-of-the-art open-source and proprietary LLMs, we establish three core findings: a) \emph{persistent limitations in structured reasoning}, with even the strongest model achieving only 32.2\% accuracy on \textsc{OneEval}\textsubscript{Hard}; b) \emph{performance consistently declines as the structural complexity of the knowledge base increases}, with accuracy dropping sharply from 53\% (textual reasoning) to 25\% (formal logic); and c) \emph{diminishing returns from extended reasoning chains}, highlighting the critical need for models to adapt reasoning depth appropriately to task complexity. We release the \textsc{OneEval} datasets, evaluation scripts, and baseline results publicly, accompanied by a leaderboard to facilitate ongoing advancements in structured knowledge reasoning.