 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDIA-CLIP: a universal representation learning framework for zero-shot DIA proteomics
Feb 02, 2026Data-independent acquisition mass spectrometry (DIA-MS) has established itself as a cornerstone of proteomic profiling and large-scale systems biology, offering unparalleled depth and reproducibility. Current DIA analysis frameworks, however, require semi-supervised training within each run for peptide-spectrum match (PSM) re-scoring. This approach is prone to overfitting and lacks generalizability across diverse species and experimental conditions. Here, we present DIA-CLIP, a pre-trained model shifting the DIA analysis paradigm from semi-supervised training to universal cross-modal representation learning. By integrating dual-encoder contrastive learning framework with encoder-decoder architecture, DIA-CLIP establishes a unified cross-modal representation for peptides and corresponding spectral features, achieving high-precision, zero-shot PSM inference. Extensive evaluations across diverse benchmarks demonstrate that DIA-CLIP consistently outperforms state-of-the-art tools, yielding up to a 45% increase in protein identification while achieving a 12% reduction in entrapment identifications. Moreover, DIA-CLIP holds immense potential for diverse practical applications, such as single-cell and spatial proteomics, where its enhanced identification depth facilitates the discovery of novel biomarkers and the elucidates of intricate cellular mechanisms.
Innovator-VL: A Multimodal Large Language Model for Scientific Discovery
Jan 27, 2026We present Innovator-VL, a scientific multimodal large language model designed to advance understanding and reasoning across diverse scientific domains while maintaining excellent performance on general vision tasks. Contrary to the trend of relying on massive domain-specific pretraining and opaque pipelines, our work demonstrates that principled training design and transparent methodology can yield strong scientific intelligence with substantially reduced data requirements. (i) First, we provide a fully transparent, end-to-end reproducible training pipeline, covering data collection, cleaning, preprocessing, supervised fine-tuning, reinforcement learning, and evaluation, along with detailed optimization recipes. This facilitates systematic extension by the community. (ii) Second, Innovator-VL exhibits remarkable data efficiency, achieving competitive performance on various scientific tasks using fewer than five million curated samples without large-scale pretraining. These results highlight that effective reasoning can be achieved through principled data selection rather than indiscriminate scaling. (iii) Third, Innovator-VL demonstrates strong generalization, achieving competitive performance on general vision, multimodal reasoning, and scientific benchmarks. This indicates that scientific alignment can be integrated into a unified model without compromising general-purpose capabilities. Our practices suggest that efficient, reproducible, and high-performing scientific multimodal models can be built even without large-scale data, providing a practical foundation for future research.
From Human Labels to Literature: Semi-Supervised Learning of NMR Chemical Shifts at Scale
Jan 26, 2026Accurate prediction of nuclear magnetic resonance (NMR) chemical shifts is fundamental to spectral analysis and molecular structure elucidation, yet existing machine learning methods rely on limited, labor-intensive atom-assigned datasets. We propose a semi-supervised framework that learns NMR chemical shifts from millions of literature-extracted spectra without explicit atom-level assignments, integrating a small amount of labeled data with large-scale unassigned spectra. We formulate chemical shift prediction from literature spectra as a permutation-invariant set supervision problem, and show that under commonly satisfied conditions on the loss function, optimal bipartite matching reduces to a sorting-based loss, enabling stable large-scale semi-supervised training beyond traditional curated datasets. Our models achieve substantially improved accuracy and robustness over state-of-the-art methods and exhibit stronger generalization on significantly larger and more diverse molecular datasets. Moreover, by incorporating solvent information at scale, our approach captures systematic solvent effects across common NMR solvents for the first time. Overall, our results demonstrate that large-scale unlabeled spectra mined from the literature can serve as a practical and effective data source for training NMR shift models, suggesting a broader role of literature-derived, weakly structured data in data-centric AI for science.
Toward Ultra-Long-Horizon Agentic Science: Cognitive Accumulation for Machine Learning Engineering
Jan 15, 2026The advancement of artificial intelligence toward agentic science is currently bottlenecked by the challenge of ultra-long-horizon autonomy, the ability to sustain strategic coherence and iterative correction over experimental cycles spanning days or weeks. While Large Language Models (LLMs) have demonstrated prowess in short-horizon reasoning, they are easily overwhelmed by execution details in the high-dimensional, delayed-feedback environments of real-world research, failing to consolidate sparse feedback into coherent long-term guidance. Here, we present ML-Master 2.0, an autonomous agent that masters ultra-long-horizon machine learning engineering (MLE) which is a representative microcosm of scientific discovery. By reframing context management as a process of cognitive accumulation, our approach introduces Hierarchical Cognitive Caching (HCC), a multi-tiered architecture inspired by computer systems that enables the structural differentiation of experience over time. By dynamically distilling transient execution traces into stable knowledge and cross-task wisdom, HCC allows agents to decouple immediate execution from long-term experimental strategy, effectively overcoming the scaling limits of static context windows. In evaluations on OpenAI's MLE-Bench under 24-hour budgets, ML-Master 2.0 achieves a state-of-the-art medal rate of 56.44%. Our findings demonstrate that ultra-long-horizon autonomy provides a scalable blueprint for AI capable of autonomous exploration beyond human-precedent complexities.
Bohrium + SciMaster: Building the Infrastructure and Ecosystem for Agentic Science at Scale
Dec 23, 2025



AI agents are emerging as a practical way to run multi-step scientific workflows that interleave reasoning with tool use and verification, pointing to a shift from isolated AI-assisted steps toward \emph{agentic science at scale}. This shift is increasingly feasible, as scientific tools and models can be invoked through stable interfaces and verified with recorded execution traces, and increasingly necessary, as AI accelerates scientific output and stresses the peer-review and publication pipeline, raising the bar for traceability and credible evaluation. However, scaling agentic science remains difficult: workflows are hard to observe and reproduce; many tools and laboratory systems are not agent-ready; execution is hard to trace and govern; and prototype AI Scientist systems are often bespoke, limiting reuse and systematic improvement from real workflow signals. We argue that scaling agentic science requires an infrastructure-and-ecosystem approach, instantiated in Bohrium+SciMaster. Bohrium acts as a managed, traceable hub for AI4S assets -- akin to a HuggingFace of AI for Science -- that turns diverse scientific data, software, compute, and laboratory systems into agent-ready capabilities. SciMaster orchestrates these capabilities into long-horizon scientific workflows, on which scientific agents can be composed and executed. Between infrastructure and orchestration, a \emph{scientific intelligence substrate} organizes reusable models, knowledge, and components into executable building blocks for workflow reasoning and action, enabling composition, auditability, and improvement through use. We demonstrate this stack with eleven representative master agents in real workflows, achieving orders-of-magnitude reductions in end-to-end scientific cycle time and generating execution-grounded signals from real workloads at multi-million scale.
PhysMaster: Building an Autonomous AI Physicist for Theoretical and Computational Physics Research
Dec 22, 2025Advances in LLMs have produced agents with knowledge and operational capabilities comparable to human scientists, suggesting potential to assist, accelerate, and automate research. However, existing studies mainly evaluate such systems on well-defined benchmarks or general tasks like literature retrieval, limiting their end-to-end problem-solving ability in open scientific scenarios. This is particularly true in physics, which is abstract, mathematically intensive, and requires integrating analytical reasoning with code-based computation. To address this, we propose PhysMaster, an LLM-based agent functioning as an autonomous theoretical and computational physicist. PhysMaster couples absract reasoning with numerical computation and leverages LANDAU, the Layered Academic Data Universe, which preserves retrieved literature, curated prior knowledge, and validated methodological traces, enhancing decision reliability and stability. It also employs an adaptive exploration strategy balancing efficiency and open-ended exploration, enabling robust performance in ultra-long-horizon tasks. We evaluate PhysMaster on problems from high-energy theory, condensed matter theory to astrophysics, including: (i) acceleration, compressing labor-intensive research from months to hours; (ii) automation, autonomously executing hypothesis-driven loops ; and (iii) autonomous discovery, independently exploring open problems.
DataFlow: An LLM-Driven Framework for Unified Data Preparation and Workflow Automation in the Era of Data-Centric AI
Dec 18, 2025The rapidly growing demand for high-quality data in Large Language Models (LLMs) has intensified the need for scalable, reliable, and semantically rich data preparation pipelines. However, current practices remain dominated by ad-hoc scripts and loosely specified workflows, which lack principled abstractions, hinder reproducibility, and offer limited support for model-in-the-loop data generation. To address these challenges, we present DataFlow, a unified and extensible LLM-driven data preparation framework. DataFlow is designed with system-level abstractions that enable modular, reusable, and composable data transformations, and provides a PyTorch-style pipeline construction API for building debuggable and optimizable dataflows. The framework consists of nearly 200 reusable operators and six domain-general pipelines spanning text, mathematical reasoning, code, Text-to-SQL, agentic RAG, and large-scale knowledge extraction. To further improve usability, we introduce DataFlow-Agent, which automatically translates natural-language specifications into executable pipelines via operator synthesis, pipeline planning, and iterative verification. Across six representative use cases, DataFlow consistently improves downstream LLM performance. Our math, code, and text pipelines outperform curated human datasets and specialized synthetic baselines, achieving up to +3\% execution accuracy in Text-to-SQL over SynSQL, +7\% average improvements on code benchmarks, and 1--3 point gains on MATH, GSM8K, and AIME. Moreover, a unified 10K-sample dataset produced by DataFlow enables base models to surpass counterparts trained on 1M Infinity-Instruct data. These results demonstrate that DataFlow provides a practical and high-performance substrate for reliable, reproducible, and scalable LLM data preparation, and establishes a system-level foundation for future data-centric AI development.
Inverse Knowledge Search over Verifiable Reasoning: Synthesizing a Scientific Encyclopedia from a Long Chains-of-Thought Knowledge Base
Oct 30, 2025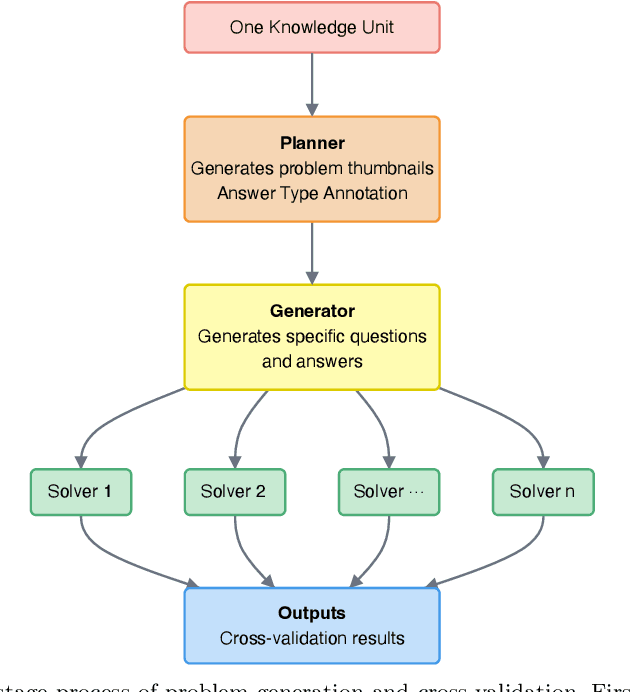
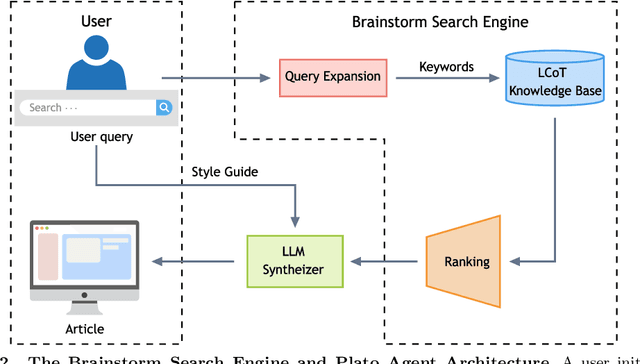
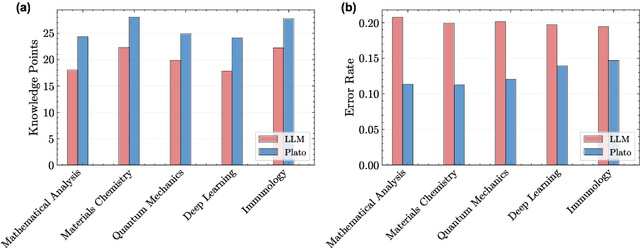
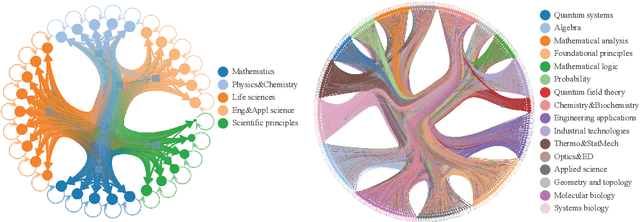
Most scientific materials compress reasoning, presenting conclusions while omitting the derivational chains that justify them. This compression hinders verification by lacking explicit, step-wise justifications and inhibits cross-domain links by collapsing the very pathways that establish the logical and causal connections between concepts. We introduce a scalable framework that decompresses scientific reasoning, constructing a verifiable Long Chain-of-Thought (LCoT) knowledge base and projecting it into an emergent encyclopedia, SciencePedia. Our pipeline operationalizes an endpoint-driven, reductionist strategy: a Socratic agent, guided by a curriculum of around 200 courses, generates approximately 3 million first-principles questions. To ensure high fidelity, multiple independent solver models generate LCoTs, which are then rigorously filtered by prompt sanitization and cross-model answer consensus, retaining only those with verifiable endpoints. This verified corpus powers the Brainstorm Search Engine, which performs inverse knowledge search -- retrieving diverse, first-principles derivations that culminate in a target concept. This engine, in turn, feeds the Plato synthesizer, which narrates these verified chains into coherent articles. The initial SciencePedia comprises approximately 200,000 fine-grained entries spanning mathematics, physics, chemistry, biology, engineering, and computation. In evaluations across six disciplines, Plato-synthesized articles (conditioned on retrieved LCoTs) exhibit substantially higher knowledge-point density and significantly lower factual error rates than an equally-prompted baseline without retrieval (as judged by an external LLM). Built on this verifiable LCoT knowledge base, this reasoning-centric approach enables trustworthy, cross-domain scientific synthesis at scale and establishes the foundation for an ever-expanding encyclopedia.
GradPower: Powering Gradients for Faster Language Model Pre-Training
May 30, 2025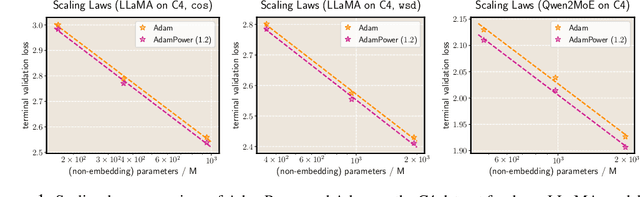
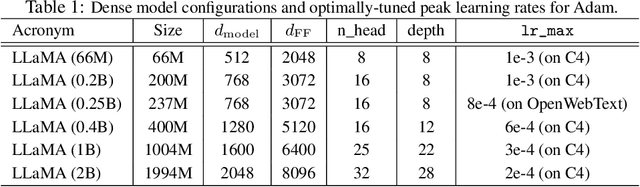
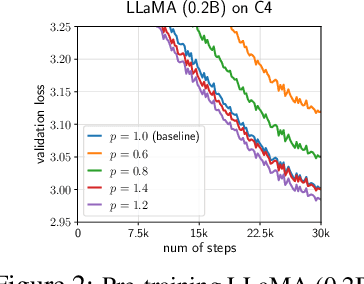
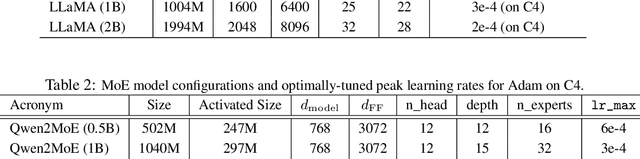
We propose GradPower, a lightweight gradient-transformation technique for accelerating language model pre-training. Given a gradient vector $g=(g_i)_i$, GradPower first applies the elementwise sign-power transformation: $\varphi_p(g)=({\rm sign}(g_i)|g_i|^p)_{i}$ for a fixed $p>0$, and then feeds the transformed gradient into a base optimizer. Notably, GradPower requires only a single-line code change and no modifications to the base optimizer's internal logic, including the hyperparameters. When applied to Adam (termed AdamPower), GradPower consistently achieves lower terminal loss across diverse architectures (LLaMA, Qwen2MoE), parameter scales (66M to 2B), datasets (C4, OpenWebText), and learning-rate schedules (cosine, warmup-stable-decay). The most pronounced gains are observed when training modern mixture-of-experts models with warmup-stable-decay schedules. GradPower also integrates seamlessly with other state-of-the-art optimizers, such as Muon, yielding further improvements. Finally, we provide theoretical analyses that reveal the underlying mechanism of GradPower and highlights the influence of gradient noise.
On the Expressive Power of Mixture-of-Experts for Structured Complex Tasks
May 30, 2025Mixture-of-experts networks (MoEs) have demonstrated remarkable efficiency in modern deep learning. Despite their empirical success, the theoretical foundations underlying their ability to model complex tasks remain poorly understood. In this work, we conduct a systematic study of the expressive power of MoEs in modeling complex tasks with two common structural priors: low-dimensionality and sparsity. For shallow MoEs, we prove that they can efficiently approximate functions supported on low-dimensional manifolds, overcoming the curse of dimensionality. For deep MoEs, we show that $\cO(L)$-layer MoEs with $E$ experts per layer can approximate piecewise functions comprising $E^L$ pieces with compositional sparsity, i.e., they can exhibit an exponential number of structured tasks. Our analysis reveals the roles of critical architectural components and hyperparameters in MoEs, including the gating mechanism, expert networks, the number of experts, and the number of layers, and offers natural suggestions for MoE variants.