 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRetrosynthesis Prediction with Local Template Retrieval
Jun 07, 2023



Retrosynthesis, which predicts the reactants of a given target molecule, is an essential task for drug discovery. In recent years, the machine learing based retrosynthesis methods have achieved promising results. In this work, we introduce RetroKNN, a local reaction template retrieval method to further boost the performance of template-based systems with non-parametric retrieval. We first build an atom-template store and a bond-template store that contain the local templates in the training data, then retrieve from these templates with a k-nearest-neighbor (KNN) search during inference. The retrieved templates are combined with neural network predictions as the final output. Furthermore, we propose a lightweight adapter to adjust the weights when combing neural network and KNN predictions conditioned on the hidden representation and the retrieved templates. We conduct comprehensive experiments on two widely used benchmarks, the USPTO-50K and USPTO-MIT. Especially for the top-1 accuracy, we improved 7.1% on the USPTO-50K dataset and 12.0% on the USPTO-MIT dataset. These results demonstrate the effectiveness of our method.
MolXPT: Wrapping Molecules with Text for Generative Pre-training
May 26, 2023



Generative pre-trained Transformer (GPT) has demonstrates its great success in natural language processing and related techniques have been adapted into molecular modeling. Considering that text is the most important record for scientific discovery, in this paper, we propose MolXPT, a unified language model of text and molecules pre-trained on SMILES (a sequence representation of molecules) wrapped by text. Briefly, we detect the molecule names in each sequence and replace them to the corresponding SMILES. In this way, the SMILES could leverage the information from surrounding text, and vice versa. The above wrapped sequences, text sequences from PubMed and SMILES sequences from PubChem are all fed into a language model for pre-training. Experimental results demonstrate that MolXPT outperforms strong baselines of molecular property prediction on MoleculeNet, performs comparably to the best model in text-molecule translation while using less than half of its parameters, and enables zero-shot molecular generation without finetuning.
What are the Desired Characteristics of Calibration Sets? Identifying Correlates on Long Form Scientific Summarization
May 12, 2023



Summarization models often generate text that is poorly calibrated to quality metrics because they are trained to maximize the likelihood of a single reference (MLE). To address this, recent work has added a calibration step, which exposes a model to its own ranked outputs to improve relevance or, in a separate line of work, contrasts positive and negative sets to improve faithfulness. While effective, much of this work has focused on how to generate and optimize these sets. Less is known about why one setup is more effective than another. In this work, we uncover the underlying characteristics of effective sets. For each training instance, we form a large, diverse pool of candidates and systematically vary the subsets used for calibration fine-tuning. Each selection strategy targets distinct aspects of the sets, such as lexical diversity or the size of the gap between positive and negatives. On three diverse scientific long-form summarization datasets (spanning biomedical, clinical, and chemical domains), we find, among others, that faithfulness calibration is optimal when the negative sets are extractive and more likely to be generated, whereas for relevance calibration, the metric margin between candidates should be maximized and surprise--the disagreement between model and metric defined candidate rankings--minimized. Code to create, select, and optimize calibration sets is available at https://github.com/griff4692/calibrating-summaries
ResiDual: Transformer with Dual Residual Connections
Apr 28, 2023



Transformer networks have become the preferred architecture for many tasks due to their state-of-the-art performance. However, the optimal way to implement residual connections in Transformer, which are essential for effective training, is still debated. Two widely used variants are the Post-Layer-Normalization (Post-LN) and Pre-Layer-Normalization (Pre-LN) Transformers, which apply layer normalization after each residual block's output or before each residual block's input, respectively. While both variants enjoy their advantages, they also suffer from severe limitations: Post-LN causes gradient vanishing issue that hinders training deep Transformers, and Pre-LN causes representation collapse issue that limits model capacity. In this paper, we propose ResiDual, a novel Transformer architecture with Pre-Post-LN (PPLN), which fuses the connections in Post-LN and Pre-LN together and inherits their advantages while avoids their limitations. We conduct both theoretical analyses and empirical experiments to verify the effectiveness of ResiDual. Theoretically, we prove that ResiDual has a lower bound on the gradient to avoid the vanishing issue due to the residual connection from Pre-LN. Moreover, ResiDual also has diverse model representations to avoid the collapse issue due to the residual connection from Post-LN. Empirically, ResiDual outperforms both Post-LN and Pre-LN on several machine translation benchmarks across different network depths and data sizes. Thanks to the good theoretical and empirical performance, ResiDual Transformer can serve as a foundation architecture for different AI models (e.g., large language models). Our code is available at https://github.com/microsoft/ResiDual.
De Novo Molecular Generation via Connection-aware Motif Mining
Feb 02, 2023



De novo molecular generation is an essential task for science discovery. Recently, fragment-based deep generative models have attracted much research attention due to their flexibility in generating novel molecules based on existing molecule fragments. However, the motif vocabulary, i.e., the collection of frequent fragments, is usually built upon heuristic rules, which brings difficulties to capturing common substructures from large amounts of molecules. In this work, we propose a new method, MiCaM, to generate molecules based on mined connection-aware motifs. Specifically, it leverages a data-driven algorithm to automatically discover motifs from a molecule library by iteratively merging subgraphs based on their frequency. The obtained motif vocabulary consists of not only molecular motifs (i.e., the frequent fragments), but also their connection information, indicating how the motifs are connected with each other. Based on the mined connection-aware motifs, MiCaM builds a connection-aware generator, which simultaneously picks up motifs and determines how they are connected. We test our method on distribution-learning benchmarks (i.e., generating novel molecules to resemble the distribution of a given training set) and goal-directed benchmarks (i.e., generating molecules with target properties), and achieve significant improvements over previous fragment-based baselines. Furthermore, we demonstrate that our method can effectively mine domain-specific motifs for different tasks.
Retrosynthetic Planning with Dual Value Networks
Jan 31, 2023Retrosynthesis, which aims to find a route to synthesize a target molecule from commercially available starting materials, is a critical task in drug discovery and materials design. Recently, the combination of ML-based single-step reaction predictors with multi-step planners has led to promising results. However, the single-step predictors are mostly trained offline to optimize the single-step accuracy, without considering complete routes. Here, we leverage reinforcement learning (RL) to improve the single-step predictor, by using a tree-shaped MDP to optimize complete routes while retaining single-step accuracy. Desirable routes should be both synthesizable and of low cost. We propose an online training algorithm, called Planning with Dual Value Networks (PDVN), in which two value networks predict the synthesizability and cost of molecules, respectively. To maintain the single-step accuracy, we design a two-branch network structure for the single-step predictor. On the widely-used USPTO dataset, our PDVN algorithm improves the search success rate of existing multi-step planners (e.g., increasing the success rate from 85.79% to 98.95% for Retro*, and reducing the number of model calls by half while solving 99.47% molecules for RetroGraph). Furthermore, PDVN finds shorter synthesis routes (e.g., reducing the average route length from 5.76 to 4.83 for Retro*, and from 5.63 to 4.78 for RetroGraph).
Incorporating Pre-training Paradigm for Antibody Sequence-Structure Co-design
Nov 17, 2022Antibodies are versatile proteins that can bind to pathogens and provide effective protection for human body. Recently, deep learning-based computational antibody design has attracted popular attention since it automatically mines the antibody patterns from data that could be complementary to human experiences. However, the computational methods heavily rely on high-quality antibody structure data, which is quite limited. Besides, the complementarity-determining region (CDR), which is the key component of an antibody that determines the specificity and binding affinity, is highly variable and hard to predict. Therefore, the data limitation issue further raises the difficulty of CDR generation for antibodies. Fortunately, there exists a large amount of sequence data of antibodies that can help model the CDR and alleviate the reliance on structure data. By witnessing the success of pre-training models for protein modeling, in this paper, we develop the antibody pre-training language model and incorporate it into the (antigen-specific) antibody design model in a systemic way. Specifically, we first pre-train an antibody language model based on the sequence data, then propose a one-shot way for sequence and structure generation of CDR to avoid the heavy cost and error propagation from an autoregressive manner, and finally leverage the pre-trained antibody model for the antigen-specific antibody generation model with some carefully designed modules. Through various experiments, we show that our method achieves superior performances over previous baselines on different tasks, such as sequence and structure generation and antigen-binding CDR-H3 design.
Unified 2D and 3D Pre-Training of Molecular Representations
Jul 14, 2022
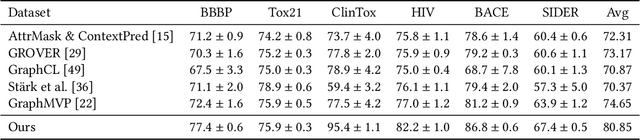
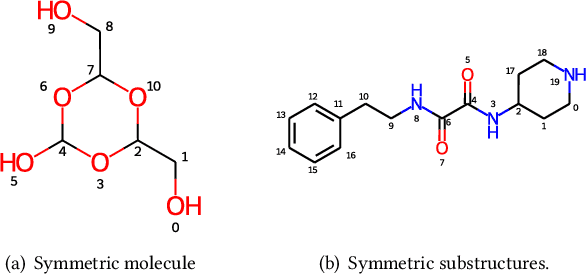
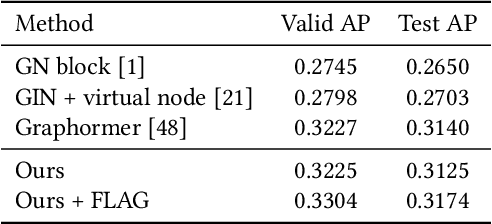
Molecular representation learning has attracted much attention recently. A molecule can be viewed as a 2D graph with nodes/atoms connected by edges/bonds, and can also be represented by a 3D conformation with 3-dimensional coordinates of all atoms. We note that most previous work handles 2D and 3D information separately, while jointly leveraging these two sources may foster a more informative representation. In this work, we explore this appealing idea and propose a new representation learning method based on a unified 2D and 3D pre-training. Atom coordinates and interatomic distances are encoded and then fused with atomic representations through graph neural networks. The model is pre-trained on three tasks: reconstruction of masked atoms and coordinates, 3D conformation generation conditioned on 2D graph, and 2D graph generation conditioned on 3D conformation. We evaluate our method on 11 downstream molecular property prediction tasks: 7 with 2D information only and 4 with both 2D and 3D information. Our method achieves state-of-the-art results on 10 tasks, and the average improvement on 2D-only tasks is 8.3%. Our method also achieves significant improvement on two 3D conformation generation tasks.
Building Multilingual Machine Translation Systems That Serve Arbitrary X-Y Translations
Jun 30, 2022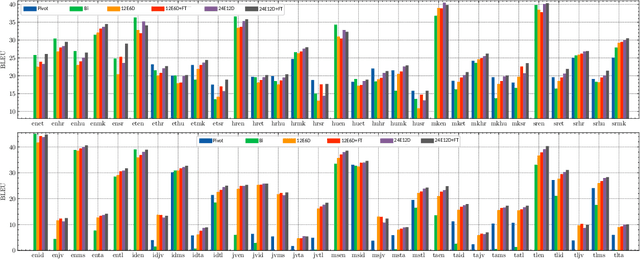
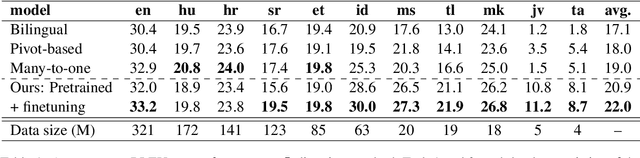
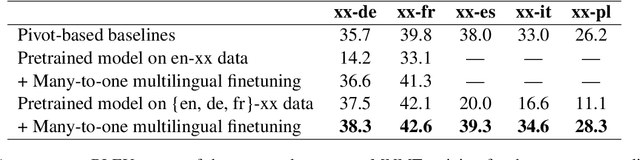
Multilingual Neural Machine Translation (MNMT) enables one system to translate sentences from multiple source languages to multiple target languages, greatly reducing deployment costs compared with conventional bilingual systems. The MNMT training benefit, however, is often limited to many-to-one directions. The model suffers from poor performance in one-to-many and many-to-many with zero-shot setup. To address this issue, this paper discusses how to practically build MNMT systems that serve arbitrary X-Y translation directions while leveraging multilinguality with a two-stage training strategy of pretraining and finetuning. Experimenting with the WMT'21 multilingual translation task, we demonstrate that our systems outperform the conventional baselines of direct bilingual models and pivot translation models for most directions, averagely giving +6.0 and +4.1 BLEU, without the need for architecture change or extra data collection. Moreover, we also examine our proposed approach in an extremely large-scale data setting to accommodate practical deployment scenarios.
RetroGraph: Retrosynthetic Planning with Graph Search
Jun 23, 2022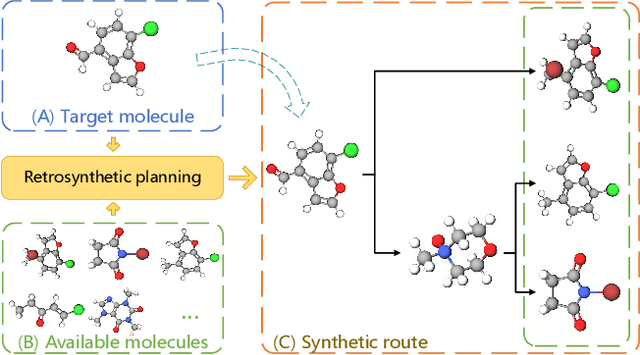
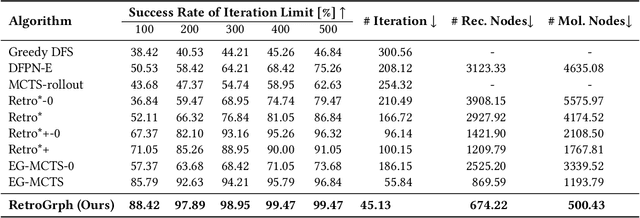
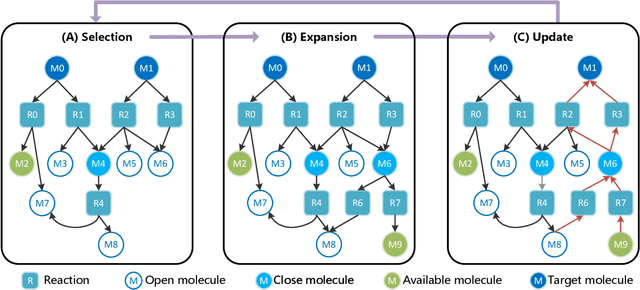
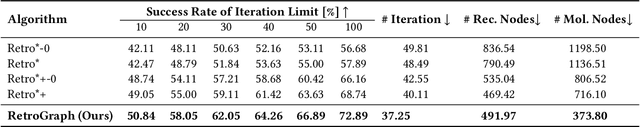
Retrosynthetic planning, which aims to find a reaction pathway to synthesize a target molecule, plays an important role in chemistry and drug discovery. This task is usually modeled as a search problem. Recently, data-driven methods have attracted many research interests and shown promising results for retrosynthetic planning. We observe that the same intermediate molecules are visited many times in the searching process, and they are usually independently treated in previous tree-based methods (e.g., AND-OR tree search, Monte Carlo tree search). Such redundancies make the search process inefficient. We propose a graph-based search policy that eliminates the redundant explorations of any intermediate molecules. As searching over a graph is more complicated than over a tree, we further adopt a graph neural network to guide the search over graphs. Meanwhile, our method can search a batch of targets together in the graph and remove the inter-target duplication in the tree-based search methods. Experimental results on two datasets demonstrate the effectiveness of our method. Especially on the widely used USPTO benchmark, we improve the search success rate to 99.47%, advancing previous state-of-the-art performance for 2.6 points.