 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConcurrent Linguistic Error Detection (CLED) for Large Language Models
Mar 25, 2024



The wide adoption of Large language models (LLMs) makes their dependability a pressing concern. Detection of errors is the first step to mitigating their impact on a system and thus, efficient error detection for LLMs is an important issue. In many settings, the LLM is considered as a black box with no access to the internal nodes; this prevents the use of many error detection schemes that need access to the model's internal nodes. An interesting observation is that the output of LLMs in error-free operation should be valid and normal text. Therefore, when the text is not valid or differs significantly from normal text, it is likely that there is an error. Based on this observation we propose to perform Concurrent Linguistic Error Detection (CLED); this scheme extracts some linguistic features of the text generated by the LLM and feeds them to a concurrent classifier that detects errors. Since the proposed error detection mechanism only relies on the outputs of the model, then it can be used on LLMs in which there is no access to the internal nodes. The proposed CLED scheme has been evaluated on the T5 model when used for news summarization and on the OPUS-MT model when used for translation. In both cases, the same set of linguistic features has been used for error detection to illustrate the applicability of the proposed scheme beyond a specific case. The results show that CLED can detect most of the errors at a low overhead penalty. The use of the concurrent classifier also enables a trade-off between error detection effectiveness and its associated overhead, so providing flexibility to a designer.
Leveraging Biomolecule and Natural Language through Multi-Modal Learning: A Survey
Mar 05, 2024



The integration of biomolecular modeling with natural language (BL) has emerged as a promising interdisciplinary area at the intersection of artificial intelligence, chemistry and biology. This approach leverages the rich, multifaceted descriptions of biomolecules contained within textual data sources to enhance our fundamental understanding and enable downstream computational tasks such as biomolecule property prediction. The fusion of the nuanced narratives expressed through natural language with the structural and functional specifics of biomolecules described via various molecular modeling techniques opens new avenues for comprehensively representing and analyzing biomolecules. By incorporating the contextual language data that surrounds biomolecules into their modeling, BL aims to capture a holistic view encompassing both the symbolic qualities conveyed through language as well as quantitative structural characteristics. In this review, we provide an extensive analysis of recent advancements achieved through cross modeling of biomolecules and natural language. (1) We begin by outlining the technical representations of biomolecules employed, including sequences, 2D graphs, and 3D structures. (2) We then examine in depth the rationale and key objectives underlying effective multi-modal integration of language and molecular data sources. (3) We subsequently survey the practical applications enabled to date in this developing research area. (4) We also compile and summarize the available resources and datasets to facilitate future work. (5) Looking ahead, we identify several promising research directions worthy of further exploration and investment to continue advancing the field. The related resources and contents are updating in \url{https://github.com/QizhiPei/Awesome-Biomolecule-Language-Cross-Modeling}.
BioT5+: Towards Generalized Biological Understanding with IUPAC Integration and Multi-task Tuning
Feb 27, 2024



Recent research trends in computational biology have increasingly focused on integrating text and bio-entity modeling, especially in the context of molecules and proteins. However, previous efforts like BioT5 faced challenges in generalizing across diverse tasks and lacked a nuanced understanding of molecular structures, particularly in their textual representations (e.g., IUPAC). This paper introduces BioT5+, an extension of the BioT5 framework, tailored to enhance biological research and drug discovery. BioT5+ incorporates several novel features: integration of IUPAC names for molecular understanding, inclusion of extensive bio-text and molecule data from sources like bioRxiv and PubChem, the multi-task instruction tuning for generality across tasks, and a novel numerical tokenization technique for improved processing of numerical data. These enhancements allow BioT5+ to bridge the gap between molecular representations and their textual descriptions, providing a more holistic understanding of biological entities, and largely improving the grounded reasoning of bio-text and bio-sequences. The model is pre-trained and fine-tuned with a large number of experiments, including \emph{3 types of problems (classification, regression, generation), 15 kinds of tasks, and 21 total benchmark datasets}, demonstrating the remarkable performance and state-of-the-art results in most cases. BioT5+ stands out for its ability to capture intricate relationships in biological data, thereby contributing significantly to bioinformatics and computational biology. Our code is available at \url{https://github.com/QizhiPei/BioT5}.
FABind: Fast and Accurate Protein-Ligand Binding
Oct 17, 2023Modeling the interaction between proteins and ligands and accurately predicting their binding structures is a critical yet challenging task in drug discovery. Recent advancements in deep learning have shown promise in addressing this challenge, with sampling-based and regression-based methods emerging as two prominent approaches. However, these methods have notable limitations. Sampling-based methods often suffer from low efficiency due to the need for generating multiple candidate structures for selection. On the other hand, regression-based methods offer fast predictions but may experience decreased accuracy. Additionally, the variation in protein sizes often requires external modules for selecting suitable binding pockets, further impacting efficiency. In this work, we propose $\mathbf{FABind}$, an end-to-end model that combines pocket prediction and docking to achieve accurate and fast protein-ligand binding. $\mathbf{FABind}$ incorporates a unique ligand-informed pocket prediction module, which is also leveraged for docking pose estimation. The model further enhances the docking process by incrementally integrating the predicted pocket to optimize protein-ligand binding, reducing discrepancies between training and inference. Through extensive experiments on benchmark datasets, our proposed $\mathbf{FABind}$ demonstrates strong advantages in terms of effectiveness and efficiency compared to existing methods. Our code is available at $\href{https://github.com/QizhiPei/FABind}{Github}$.
BioT5: Enriching Cross-modal Integration in Biology with Chemical Knowledge and Natural Language Associations
Oct 17, 2023



Recent advancements in biological research leverage the integration of molecules, proteins, and natural language to enhance drug discovery. However, current models exhibit several limitations, such as the generation of invalid molecular SMILES, underutilization of contextual information, and equal treatment of structured and unstructured knowledge. To address these issues, we propose $\mathbf{BioT5}$, a comprehensive pre-training framework that enriches cross-modal integration in biology with chemical knowledge and natural language associations. $\mathbf{BioT5}$ utilizes SELFIES for $100%$ robust molecular representations and extracts knowledge from the surrounding context of bio-entities in unstructured biological literature. Furthermore, $\mathbf{BioT5}$ distinguishes between structured and unstructured knowledge, leading to more effective utilization of information. After fine-tuning, BioT5 shows superior performance across a wide range of tasks, demonstrating its strong capability of capturing underlying relations and properties of bio-entities. Our code is available at $\href{https://github.com/QizhiPei/BioT5}{Github}$.
Incorporating Pre-training Paradigm for Antibody Sequence-Structure Co-design
Nov 17, 2022Antibodies are versatile proteins that can bind to pathogens and provide effective protection for human body. Recently, deep learning-based computational antibody design has attracted popular attention since it automatically mines the antibody patterns from data that could be complementary to human experiences. However, the computational methods heavily rely on high-quality antibody structure data, which is quite limited. Besides, the complementarity-determining region (CDR), which is the key component of an antibody that determines the specificity and binding affinity, is highly variable and hard to predict. Therefore, the data limitation issue further raises the difficulty of CDR generation for antibodies. Fortunately, there exists a large amount of sequence data of antibodies that can help model the CDR and alleviate the reliance on structure data. By witnessing the success of pre-training models for protein modeling, in this paper, we develop the antibody pre-training language model and incorporate it into the (antigen-specific) antibody design model in a systemic way. Specifically, we first pre-train an antibody language model based on the sequence data, then propose a one-shot way for sequence and structure generation of CDR to avoid the heavy cost and error propagation from an autoregressive manner, and finally leverage the pre-trained antibody model for the antigen-specific antibody generation model with some carefully designed modules. Through various experiments, we show that our method achieves superior performances over previous baselines on different tasks, such as sequence and structure generation and antigen-binding CDR-H3 design.
Unified 2D and 3D Pre-Training of Molecular Representations
Jul 14, 2022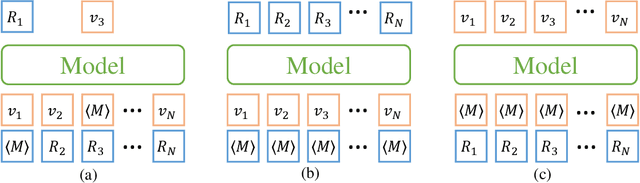
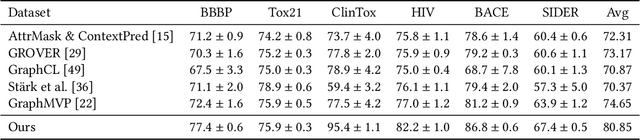
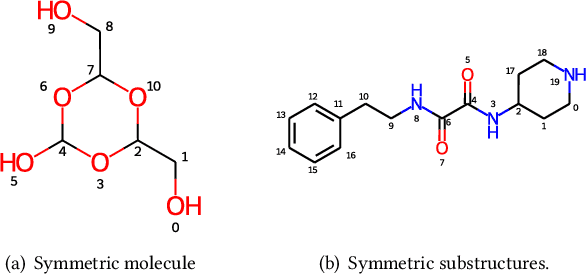
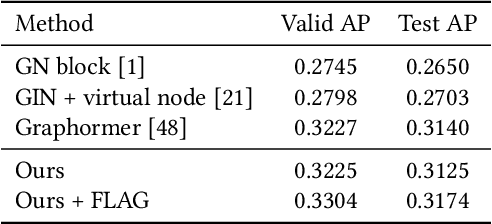
Molecular representation learning has attracted much attention recently. A molecule can be viewed as a 2D graph with nodes/atoms connected by edges/bonds, and can also be represented by a 3D conformation with 3-dimensional coordinates of all atoms. We note that most previous work handles 2D and 3D information separately, while jointly leveraging these two sources may foster a more informative representation. In this work, we explore this appealing idea and propose a new representation learning method based on a unified 2D and 3D pre-training. Atom coordinates and interatomic distances are encoded and then fused with atomic representations through graph neural networks. The model is pre-trained on three tasks: reconstruction of masked atoms and coordinates, 3D conformation generation conditioned on 2D graph, and 2D graph generation conditioned on 3D conformation. We evaluate our method on 11 downstream molecular property prediction tasks: 7 with 2D information only and 4 with both 2D and 3D information. Our method achieves state-of-the-art results on 10 tasks, and the average improvement on 2D-only tasks is 8.3%. Our method also achieves significant improvement on two 3D conformation generation tasks.
SMT-DTA: Improving Drug-Target Affinity Prediction with Semi-supervised Multi-task Training
Jun 22, 2022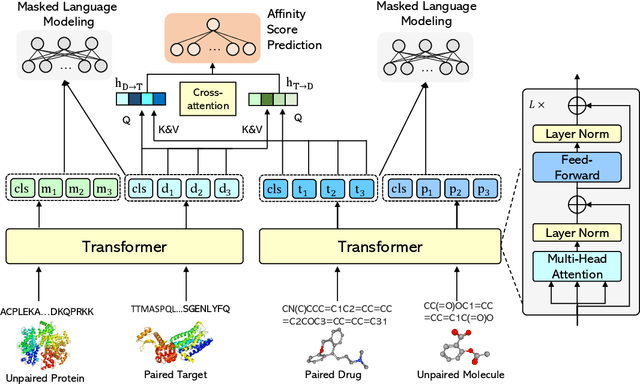
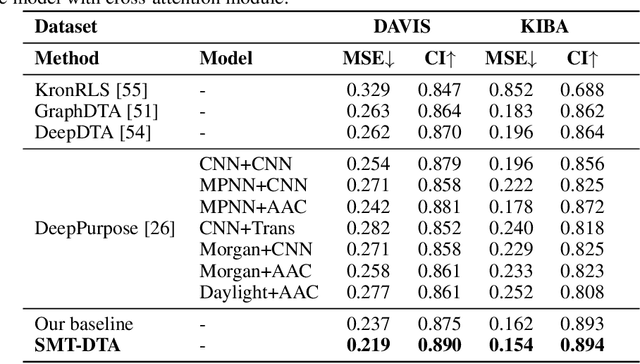
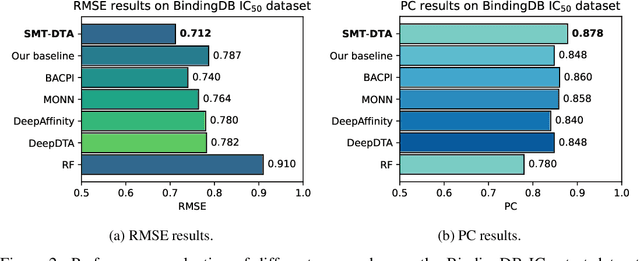

Drug-Target Affinity (DTA) prediction is an essential task for drug discovery and pharmaceutical research. Accurate predictions of DTA can greatly benefit the design of new drug. As wet experiments are costly and time consuming, the supervised data for DTA prediction is extremely limited. This seriously hinders the application of deep learning based methods, which require a large scale of supervised data. To address this challenge and improve the DTA prediction accuracy, we propose a framework with several simple yet effective strategies in this work: (1) a multi-task training strategy, which takes the DTA prediction and the masked language modeling (MLM) task on the paired drug-target dataset; (2) a semi-supervised training method to empower the drug and target representation learning by leveraging large-scale unpaired molecules and proteins in training, which differs from previous pre-training and fine-tuning methods that only utilize molecules or proteins in pre-training; and (3) a cross-attention module to enhance the interaction between drug and target representation. Extensive experiments are conducted on three real-world benchmark datasets: BindingDB, DAVIS and KIBA. The results show that our framework significantly outperforms existing methods and achieves state-of-the-art performances, e.g., $0.712$ RMSE on BindingDB IC$_{50}$ measurement with more than $5\%$ improvement than previous best work. In addition, case studies on specific drug-target binding activities, drug feature visualizations, and real-world applications demonstrate the great potential of our work. The code and data are released at https://github.com/QizhiPei/SMT-DTA
Direct Molecular Conformation Generation
Feb 03, 2022
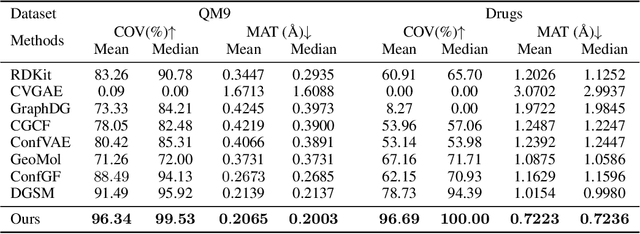
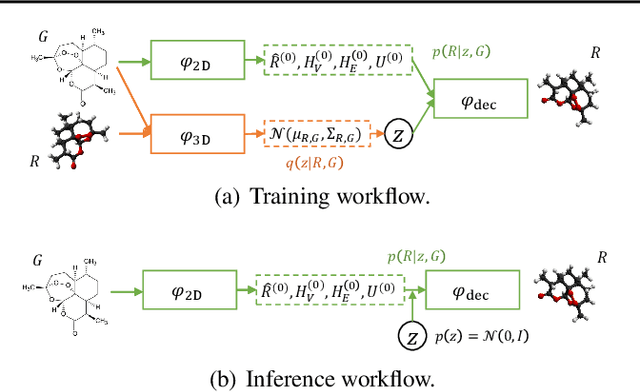
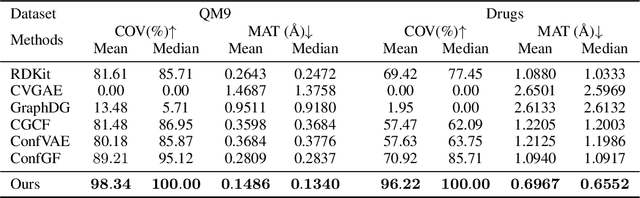
Molecular conformation generation aims to generate three-dimensional coordinates of all the atoms in a molecule and is an important task in bioinformatics and pharmacology. Previous distance-based methods first predict interatomic distances and then generate conformations based on them, which could result in conflicting distances. In this work, we propose a method that directly predicts the coordinates of atoms. We design a dedicated loss function for conformation generation, which is invariant to roto-translation of coordinates of conformations and permutation of symmetric atoms in molecules. We further design a backbone model that stacks multiple blocks, where each block refines the conformation generated by its preceding block. Our method achieves state-of-the-art results on four public benchmarks: on small-scale GEOM-QM9 and GEOM-Drugs which have $200$K training data, we can improve the previous best matching score by $3.5\%$ and $28.9\%$; on large-scale GEOM-QM9 and GEOM-Drugs which have millions of training data, those two improvements are $47.1\%$ and $36.3\%$. This shows the effectiveness of our method and the great potential of the direct approach. Our code is released at \url{https://github.com/DirectMolecularConfGen/DMCG}.
Discovering Drug-Target Interaction Knowledge from Biomedical Literature
Sep 27, 2021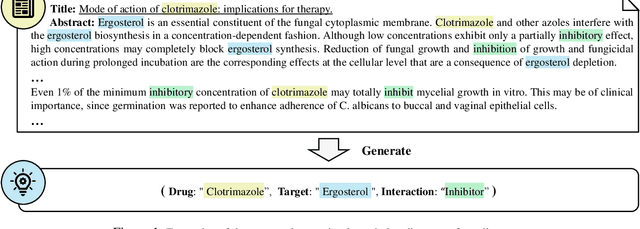

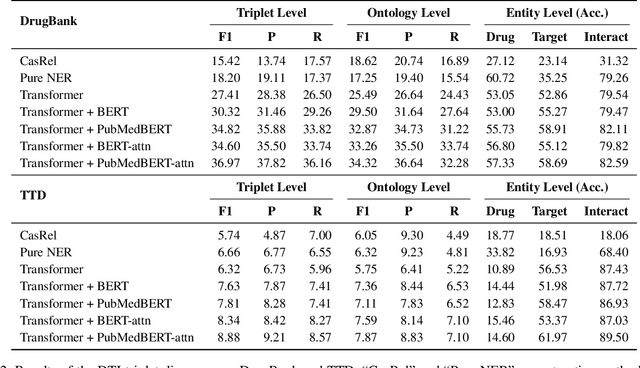
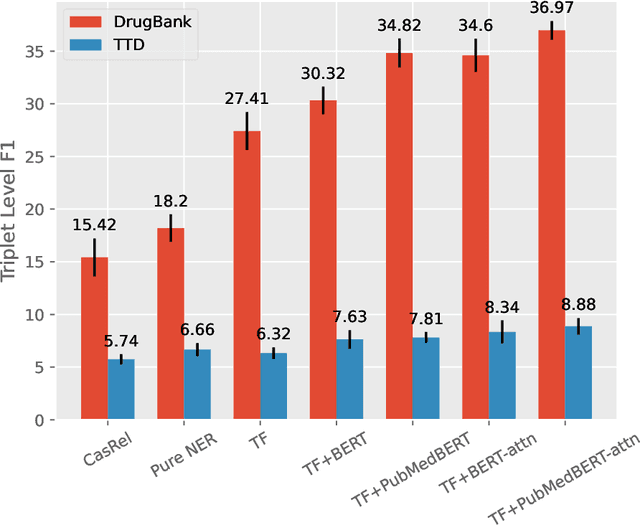
The Interaction between Drugs and Targets (DTI) in human body plays a crucial role in biomedical science and applications. As millions of papers come out every year in the biomedical domain, automatically discovering DTI knowledge from biomedical literature, which are usually triplets about drugs, targets and their interaction, becomes an urgent demand in the industry. Existing methods of discovering biological knowledge are mainly extractive approaches that often require detailed annotations (e.g., all mentions of biological entities, relations between every two entity mentions, etc.). However, it is difficult and costly to obtain sufficient annotations due to the requirement of expert knowledge from biomedical domains. To overcome these difficulties, we explore the first end-to-end solution for this task by using generative approaches. We regard the DTI triplets as a sequence and use a Transformer-based model to directly generate them without using the detailed annotations of entities and relations. Further, we propose a semi-supervised method, which leverages the aforementioned end-to-end model to filter unlabeled literature and label them. Experimental results show that our method significantly outperforms extractive baselines on DTI discovery. We also create a dataset, KD-DTI, to advance this task and will release it to the community.