 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnd-to-End Reverse Screening Identifies Protein Targets of Small Molecules Using HelixFold3
Jan 20, 2026Identifying protein targets for small molecules, or reverse screening, is essential for understanding drug action, guiding compound repurposing, predicting off-target effects, and elucidating the molecular mechanisms of bioactive compounds. Despite its critical role, reverse screening remains challenging because accurately capturing interactions between a small molecule and structurally diverse proteins is inherently complex, and conventional step-wise workflows often propagate errors across decoupled steps such as target structure modeling, pocket identification, docking, and scoring. Here, we present an end-to-end reverse screening strategy leveraging HelixFold3, a high-accuracy biomolecular structure prediction model akin to AlphaFold3, which simultaneously models the folding of proteins from a protein library and the docking of small-molecule ligands within a unified framework. We validate this approach on a diverse and representative set of approximately one hundred small molecules. Compared with conventional reverse docking, our method improves screening accuracy and demonstrates enhanced structural fidelity, binding-site precision, and target prioritization. By systematically linking small molecules to their protein targets, this framework establishes a scalable and straightforward platform for dissecting molecular mechanisms, exploring off-target interactions, and supporting rational drug discovery.
HelixDesign-Antibody: A Scalable Production-Grade Platform for Antibody Design Built on HelixFold3
Jul 03, 2025Antibody engineering is essential for developing therapeutics and advancing biomedical research. Traditional discovery methods often rely on time-consuming and resource-intensive experimental screening. To enhance and streamline this process, we introduce a production-grade, high-throughput platform built on HelixFold3, HelixDesign-Antibody, which utilizes the high-accuracy structure prediction model, HelixFold3. The platform facilitates the large-scale generation of antibody candidate sequences and evaluates their interaction with antigens. Integrated high-performance computing (HPC) support enables high-throughput screening, addressing challenges such as fragmented toolchains and high computational demands. Validation on multiple antigens showcases the platform's ability to generate diverse and high-quality antibodies, confirming a scaling law where exploring larger sequence spaces increases the likelihood of identifying optimal binders. This platform provides a seamless, accessible solution for large-scale antibody design and is available via the antibody design page of PaddleHelix platform.
HelixDesign-Binder: A Scalable Production-Grade Platform for Binder Design Built on HelixFold3
May 28, 2025Protein binder design is central to therapeutics, diagnostics, and synthetic biology, yet practical deployment remains challenging due to fragmented workflows, high computational costs, and complex tool integration. We present HelixDesign-Binder, a production-grade, high-throughput platform built on HelixFold3 that automates the full binder design pipeline, from backbone generation and sequence design to structural evaluation and multi-dimensional scoring. By unifying these stages into a scalable and user-friendly system, HelixDesign-Binder enables efficient exploration of binder candidates with favorable structural, energetic, and physicochemical properties. The platform leverages Baidu Cloud's high-performance infrastructure to support large-scale design and incorporates advanced scoring metrics, including ipTM, predicted binding free energy, and interface hydrophobicity. Benchmarking across six protein targets demonstrates that HelixDesign-Binder reliably produces diverse and high-quality binders, some of which match or exceed validated designs in predicted binding affinity. HelixDesign-Binder is accessible via an interactive web interface in PaddleHelix platform, supporting both academic research and industrial applications in antibody and protein binder development.
Precise Antigen-Antibody Structure Predictions Enhance Antibody Development with HelixFold-Multimer
Dec 13, 2024



The accurate prediction of antigen-antibody structures is essential for advancing immunology and therapeutic development, as it helps elucidate molecular interactions that underlie immune responses. Despite recent progress with deep learning models like AlphaFold and RoseTTAFold, accurately modeling antigen-antibody complexes remains a challenge due to their unique evolutionary characteristics. HelixFold-Multimer, a specialized model developed for this purpose, builds on the framework of AlphaFold-Multimer and demonstrates improved precision for antigen-antibody structures. HelixFold-Multimer not only surpasses other models in accuracy but also provides essential insights into antibody development, enabling more precise identification of binding sites, improved interaction prediction, and enhanced design of therapeutic antibodies. These advances underscore HelixFold-Multimer's potential in supporting antibody research and therapeutic innovation.
Technical Report of HelixFold3 for Biomolecular Structure Prediction
Aug 30, 2024



The AlphaFold series has transformed protein structure prediction with remarkable accuracy, often matching experimental methods. AlphaFold2, AlphaFold-Multimer, and the latest AlphaFold3 represent significant strides in predicting single protein chains, protein complexes, and biomolecular structures. While AlphaFold2 and AlphaFold-Multimer are open-sourced, facilitating rapid and reliable predictions, AlphaFold3 remains partially accessible through a limited online server and has not been open-sourced, restricting further development. To address these challenges, the PaddleHelix team is developing HelixFold3, aiming to replicate AlphaFold3's capabilities. Using insights from previous models and extensive datasets, HelixFold3 achieves an accuracy comparable to AlphaFold3 in predicting the structures of conventional ligands, nucleic acids, and proteins. The initial release of HelixFold3 is available as open source on GitHub for academic research, promising to advance biomolecular research and accelerate discoveries. We also provide online service at PaddleHelix website at https://paddlehelix.baidu.com/app/all/helixfold3/forecast.
Unifying Sequences, Structures, and Descriptions for Any-to-Any Protein Generation with the Large Multimodal Model HelixProtX
Jul 12, 2024



Proteins are fundamental components of biological systems and can be represented through various modalities, including sequences, structures, and textual descriptions. Despite the advances in deep learning and scientific large language models (LLMs) for protein research, current methodologies predominantly focus on limited specialized tasks -- often predicting one protein modality from another. These approaches restrict the understanding and generation of multimodal protein data. In contrast, large multimodal models have demonstrated potential capabilities in generating any-to-any content like text, images, and videos, thus enriching user interactions across various domains. Integrating these multimodal model technologies into protein research offers significant promise by potentially transforming how proteins are studied. To this end, we introduce HelixProtX, a system built upon the large multimodal model, aiming to offer a comprehensive solution to protein research by supporting any-to-any protein modality generation. Unlike existing methods, it allows for the transformation of any input protein modality into any desired protein modality. The experimental results affirm the advanced capabilities of HelixProtX, not only in generating functional descriptions from amino acid sequences but also in executing critical tasks such as designing protein sequences and structures from textual descriptions. Preliminary findings indicate that HelixProtX consistently achieves superior accuracy across a range of protein-related tasks, outperforming existing state-of-the-art models. By integrating multimodal large models into protein research, HelixProtX opens new avenues for understanding protein biology, thereby promising to accelerate scientific discovery.
HelixFold-Multimer: Elevating Protein Complex Structure Prediction to New Heights
Apr 16, 2024



While monomer protein structure prediction tools boast impressive accuracy, the prediction of protein complex structures remains a daunting challenge in the field. This challenge is particularly pronounced in scenarios involving complexes with protein chains from different species, such as antigen-antibody interactions, where accuracy often falls short. Limited by the accuracy of complex prediction, tasks based on precise protein-protein interaction analysis also face obstacles. In this report, we highlight the ongoing advancements of our protein complex structure prediction model, HelixFold-Multimer, underscoring its enhanced performance. HelixFold-Multimer provides precise predictions for diverse protein complex structures, especially in therapeutic protein interactions. Notably, HelixFold-Multimer achieves remarkable success in antigen-antibody and peptide-protein structure prediction, surpassing AlphaFold-Multimer by several folds. HelixFold-Multimer is now available for public use on the PaddleHelix platform, offering both a general version and an antigen-antibody version. Researchers can conveniently access and utilize this service for their development needs.
Pre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Oct 21, 2023Molecular docking, a pivotal computational tool for drug discovery, predicts the binding interactions between small molecules (ligands) and target proteins (receptors). Conventional physics-based docking tools, though widely used, face limitations in precision due to restricted conformational sampling and imprecise scoring functions. Recent endeavors have employed deep learning techniques to enhance docking accuracy, but their generalization remains a concern due to limited training data. Leveraging the success of extensive and diverse data in other domains, we introduce HelixDock, a novel approach for site-specific molecular docking. Hundreds of millions of binding poses are generated by traditional docking tools, encompassing diverse protein targets and small molecules. Our deep learning-based docking model, a SE(3)-equivariant network, is pre-trained with this large-scale dataset and then fine-tuned with a small number of precise receptor-ligand complex structures. Comparative analyses against physics-based and deep learning-based baseline methods highlight HelixDock's superiority, especially on challenging test sets. Our study elucidates the scaling laws of the pre-trained molecular docking models, showcasing consistent improvements with increased model parameters and pre-train data quantities. Harnessing the power of extensive and diverse generated data holds promise for advancing AI-driven drug discovery.
GEM-2: Next Generation Molecular Property Prediction Network with Many-body and Full-range Interaction Modeling
Aug 15, 2022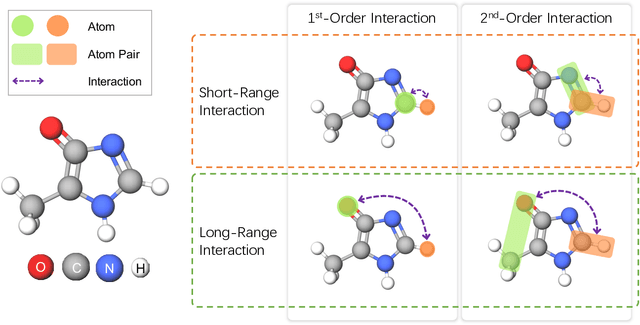
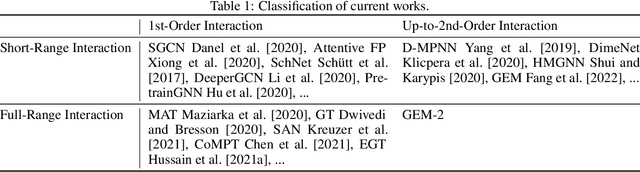
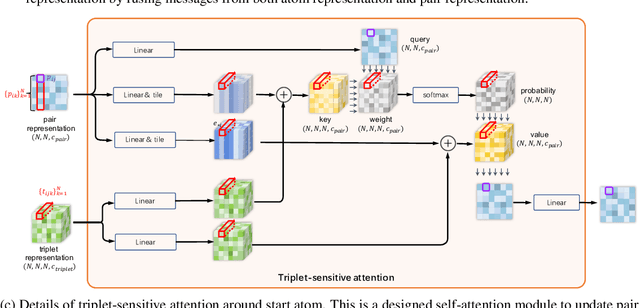
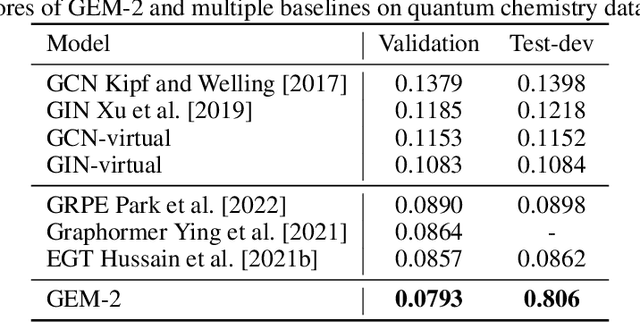
Molecular property prediction is a fundamental task in the drug and material industries. Physically, the properties of a molecule are determined by its own electronic structure, which can be exactly described by the Schr\"odinger equation. However, solving the Schr\"odinger equation for most molecules is extremely challenging due to long-range interactions in the behavior of a quantum many-body system. While deep learning methods have proven to be effective in molecular property prediction, we design a novel method, namely GEM-2, which comprehensively considers both the long-range and many-body interactions in molecules. GEM-2 consists of two interacted tracks: an atom-level track modeling both the local and global correlation between any two atoms, and a pair-level track modeling the correlation between all atom pairs, which embed information between any 3 or 4 atoms. Extensive experiments demonstrated the superiority of GEM-2 over multiple baseline methods in quantum chemistry and drug discovery tasks.
DuETA: Traffic Congestion Propagation Pattern Modeling via Efficient Graph Learning for ETA Prediction at Baidu Maps
Aug 15, 2022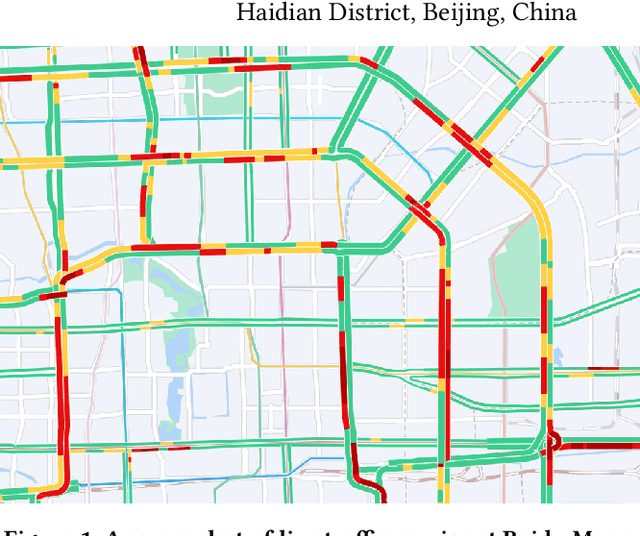
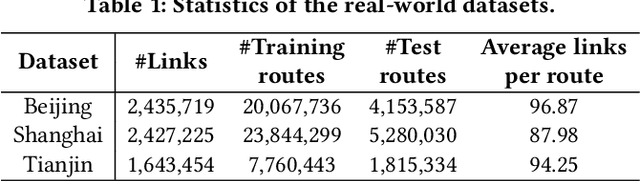
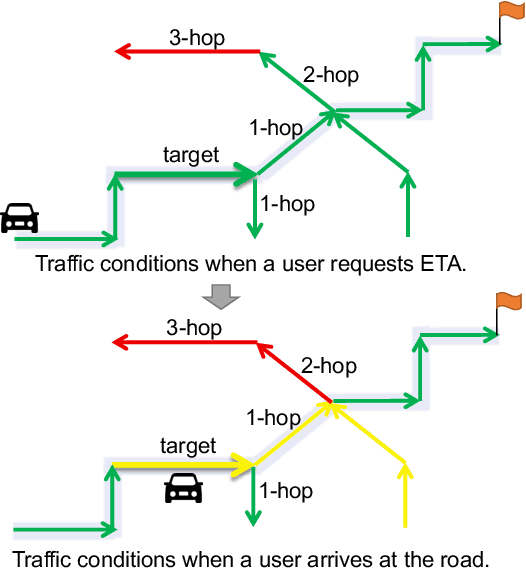
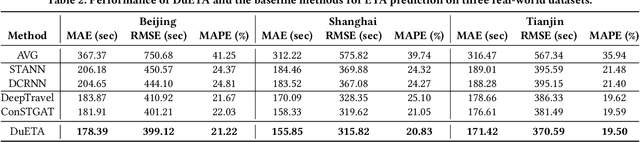
Estimated time of arrival (ETA) prediction, also known as travel time estimation, is a fundamental task for a wide range of intelligent transportation applications, such as navigation, route planning, and ride-hailing services. To accurately predict the travel time of a route, it is essential to take into account both contextual and predictive factors, such as spatial-temporal interaction, driving behavior, and traffic congestion propagation inference. The ETA prediction models previously deployed at Baidu Maps have addressed the factors of spatial-temporal interaction (ConSTGAT) and driving behavior (SSML). In this work, we focus on modeling traffic congestion propagation patterns to improve ETA performance. Traffic congestion propagation pattern modeling is challenging, and it requires accounting for impact regions over time and cumulative effect of delay variations over time caused by traffic events on the road network. In this paper, we present a practical industrial-grade ETA prediction framework named DuETA. Specifically, we construct a congestion-sensitive graph based on the correlations of traffic patterns, and we develop a route-aware graph transformer to directly learn the long-distance correlations of the road segments. This design enables DuETA to capture the interactions between the road segment pairs that are spatially distant but highly correlated with traffic conditions. Extensive experiments are conducted on large-scale, real-world datasets collected from Baidu Maps. Experimental results show that ETA prediction can significantly benefit from the learned traffic congestion propagation patterns. In addition, DuETA has already been deployed in production at Baidu Maps, serving billions of requests every day. This demonstrates that DuETA is an industrial-grade and robust solution for large-scale ETA prediction services.