 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-modal Molecule Structure-text Model for Text-based Retrieval and Editing
Dec 21, 2022There is increasing adoption of artificial intelligence in drug discovery. However, existing works use machine learning to mainly utilize the chemical structures of molecules yet ignore the vast textual knowledge available in chemistry. Incorporating textual knowledge enables us to realize new drug design objectives, adapt to text-based instructions, and predict complex biological activities. We present a multi-modal molecule structure-text model, MoleculeSTM, by jointly learning molecule's chemical structures and textual descriptions via a contrastive learning strategy. To train MoleculeSTM, we construct the largest multi-modal dataset to date, namely PubChemSTM, with over 280K chemical structure-text pairs. To demonstrate the effectiveness and utility of MoleculeSTM, we design two challenging zero-shot tasks based on text instructions, including structure-text retrieval and molecule editing. MoleculeSTM possesses two main properties: open vocabulary and compositionality via natural language. In experiments, MoleculeSTM obtains the state-of-the-art generalization ability to novel biochemical concepts across various benchmarks.
GAUCHE: A Library for Gaussian Processes in Chemistry
Dec 06, 2022We introduce GAUCHE, a library for GAUssian processes in CHEmistry. Gaussian processes have long been a cornerstone of probabilistic machine learning, affording particular advantages for uncertainty quantification and Bayesian optimisation. Extending Gaussian processes to chemical representations, however, is nontrivial, necessitating kernels defined over structured inputs such as graphs, strings and bit vectors. By defining such kernels in GAUCHE, we seek to open the door to powerful tools for uncertainty quantification and Bayesian optimisation in chemistry. Motivated by scenarios frequently encountered in experimental chemistry, we showcase applications for GAUCHE in molecular discovery and chemical reaction optimisation. The codebase is made available at https://github.com/leojklarner/gauche
EurNet: Efficient Multi-Range Relational Modeling of Spatial Multi-Relational Data
Nov 23, 2022Modeling spatial relationship in the data remains critical across many different tasks, such as image classification, semantic segmentation and protein structure understanding. Previous works often use a unified solution like relative positional encoding. However, there exists different kinds of spatial relations, including short-range, medium-range and long-range relations, and modeling them separately can better capture the focus of different tasks on the multi-range relations (e.g., short-range relations can be important in instance segmentation, while long-range relations should be upweighted for semantic segmentation). In this work, we introduce the EurNet for Efficient multi-range relational modeling. EurNet constructs the multi-relational graph, where each type of edge corresponds to short-, medium- or long-range spatial interactions. In the constructed graph, EurNet adopts a novel modeling layer, called gated relational message passing (GRMP), to propagate multi-relational information across the data. GRMP captures multiple relations within the data with little extra computational cost. We study EurNets in two important domains for image and protein structure modeling. Extensive experiments on ImageNet classification, COCO object detection and ADE20K semantic segmentation verify the gains of EurNet over the previous SoTA FocalNet. On the EC and GO protein function prediction benchmarks, EurNet consistently surpasses the previous SoTA GearNet. Our results demonstrate the strength of EurNets on modeling spatial multi-relational data from various domains. The implementations of EurNet for image modeling are available at https://github.com/hirl-team/EurNet-Image . The implementations for other applied domains/tasks will be released soon.
Flaky Performances when Pretraining on Relational Databases
Nov 09, 2022We explore the downstream task performances for graph neural network (GNN) self-supervised learning (SSL) methods trained on subgraphs extracted from relational databases (RDBs). Intuitively, this joint use of SSL and GNNs should allow to leverage more of the available data, which could translate to better results. However, we found that naively porting contrastive SSL techniques can cause ``negative transfer'': linear evaluation on fixed representations from a pretrained model performs worse than on representations from the randomly-initialized model. Based on the conjecture that contrastive SSL conflicts with the message passing layers of the GNN, we propose InfoNode: a contrastive loss aiming to maximize the mutual information between a node's initial- and final-layer representation. The primary empirical results support our conjecture and the effectiveness of InfoNode.
Learning on Large-scale Text-attributed Graphs via Variational Inference
Oct 26, 2022



This paper studies learning on text-attributed graphs (TAGs), where each node is associated with a text description. An ideal solution for such a problem would be integrating both the text and graph structure information with large language models and graph neural networks (GNNs). However, the problem becomes very challenging when graphs are large due to the high computational complexity brought by large language models and training GNNs on big graphs. In this paper, we propose an efficient and effective solution to learning on large text-attributed graphs by fusing graph structure and language learning with a variational Expectation-Maximization (EM) framework, called GLEM. Instead of simultaneously training large language models and GNNs on big graphs, GLEM proposes to alternatively update the two modules in the E-step and M-step. Such a procedure allows to separately train the two modules but at the same time allows the two modules to interact and mutually enhance each other. Extensive experiments on multiple data sets demonstrate the efficiency and effectiveness of the proposed approach.
Protein Sequence and Structure Co-Design with Equivariant Translation
Oct 17, 2022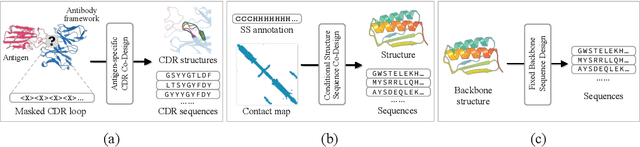
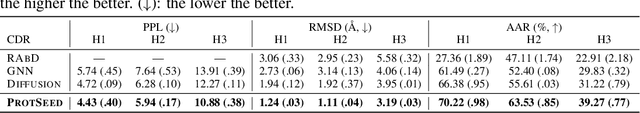
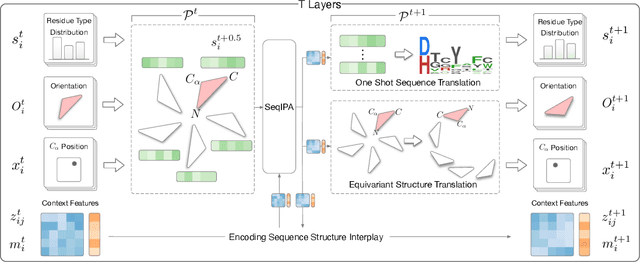
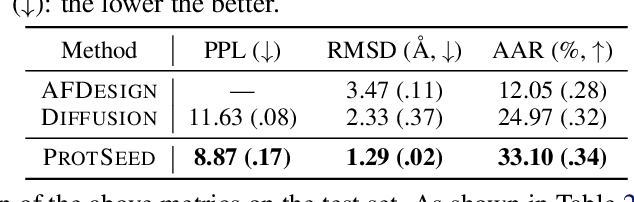
Proteins are macromolecules that perform essential functions in all living organisms. Designing novel proteins with specific structures and desired functions has been a long-standing challenge in the field of bioengineering. Existing approaches generate both protein sequence and structure using either autoregressive models or diffusion models, both of which suffer from high inference costs. In this paper, we propose a new approach capable of protein sequence and structure co-design, which iteratively translates both protein sequence and structure into the desired state from random initialization, based on context features given a priori. Our model consists of a trigonometry-aware encoder that reasons geometrical constraints and interactions from context features, and a roto-translation equivariant decoder that translates protein sequence and structure interdependently. Notably, all protein amino acids are updated in one shot in each translation step, which significantly accelerates the inference process. Experimental results across multiple tasks show that our model outperforms previous state-of-the-art baselines by a large margin, and is able to design proteins of high fidelity as regards both sequence and structure, with running time orders of magnitude less than sampling-based methods.
Inductive Logical Query Answering in Knowledge Graphs
Oct 13, 2022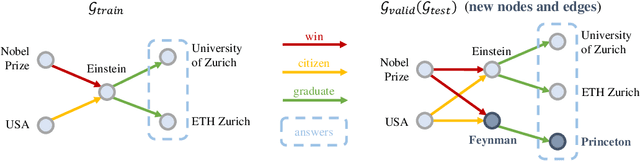
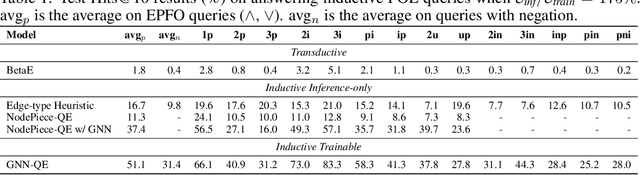
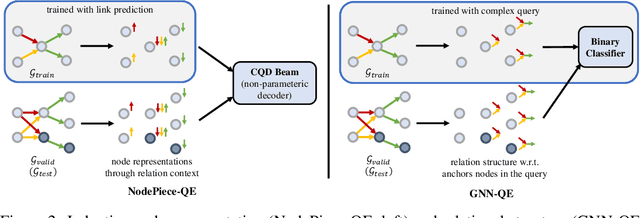
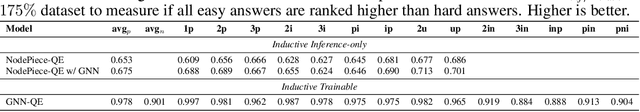
Formulating and answering logical queries is a standard communication interface for knowledge graphs (KGs). Alleviating the notorious incompleteness of real-world KGs, neural methods achieved impressive results in link prediction and complex query answering tasks by learning representations of entities, relations, and queries. Still, most existing query answering methods rely on transductive entity embeddings and cannot generalize to KGs containing new entities without retraining the entity embeddings. In this work, we study the inductive query answering task where inference is performed on a graph containing new entities with queries over both seen and unseen entities. To this end, we devise two mechanisms leveraging inductive node and relational structure representations powered by graph neural networks (GNNs). Experimentally, we show that inductive models are able to perform logical reasoning at inference time over unseen nodes generalizing to graphs up to 500% larger than training ones. Exploring the efficiency--effectiveness trade-off, we find the inductive relational structure representation method generally achieves higher performance, while the inductive node representation method is able to answer complex queries in the inference-only regime without any training on queries and scales to graphs of millions of nodes. Code is available at https://github.com/DeepGraphLearning/InductiveQE.
E3Bind: An End-to-End Equivariant Network for Protein-Ligand Docking
Oct 12, 2022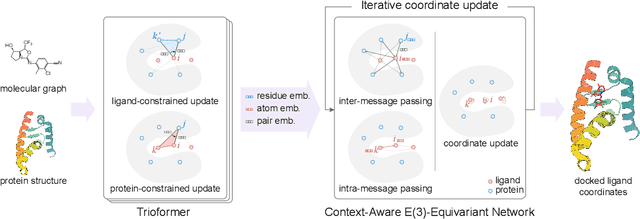
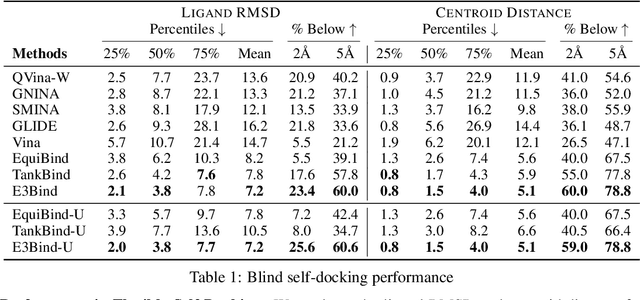
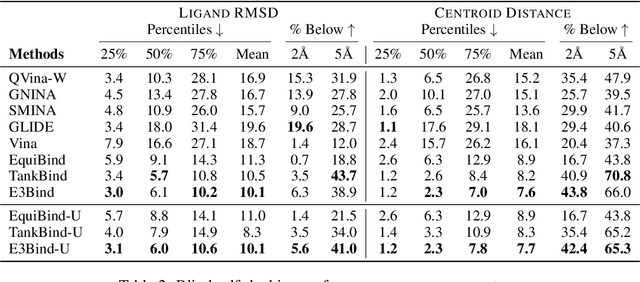
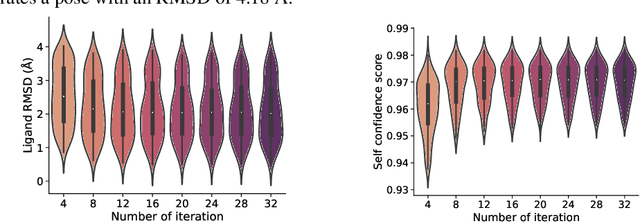
In silico prediction of the ligand binding pose to a given protein target is a crucial but challenging task in drug discovery. This work focuses on blind flexible selfdocking, where we aim to predict the positions, orientations and conformations of docked molecules. Traditional physics-based methods usually suffer from inaccurate scoring functions and high inference cost. Recently, data-driven methods based on deep learning techniques are attracting growing interest thanks to their efficiency during inference and promising performance. These methods usually either adopt a two-stage approach by first predicting the distances between proteins and ligands and then generating the final coordinates based on the predicted distances, or directly predicting the global roto-translation of ligands. In this paper, we take a different route. Inspired by the resounding success of AlphaFold2 for protein structure prediction, we propose E3Bind, an end-to-end equivariant network that iteratively updates the ligand pose. E3Bind models the protein-ligand interaction through careful consideration of the geometric constraints in docking and the local context of the binding site. Experiments on standard benchmark datasets demonstrate the superior performance of our end-to-end trainable model compared to traditional and recently-proposed deep learning methods.
Metro: Memory-Enhanced Transformer for Retrosynthetic Planning via Reaction Tree
Sep 30, 2022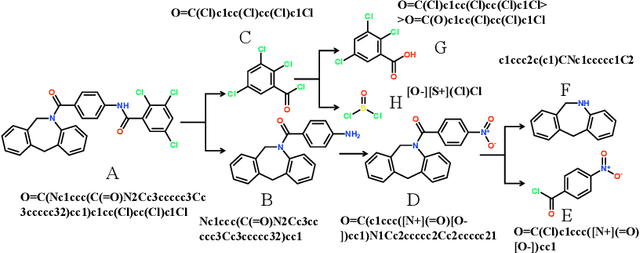
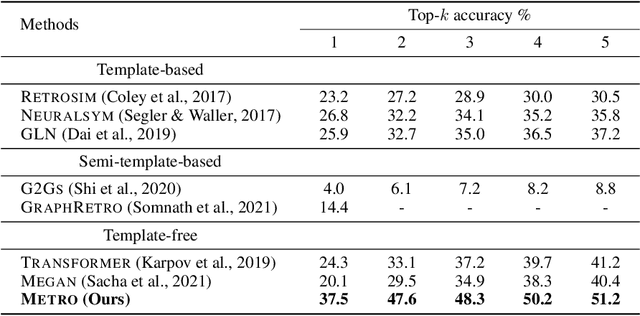
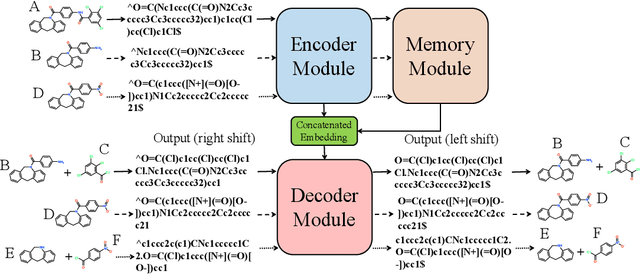
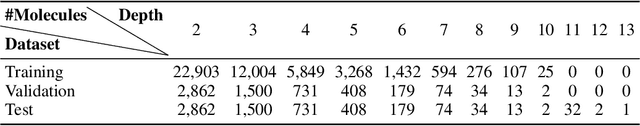
Retrosynthetic planning plays a critical role in drug discovery and organic chemistry. Starting from a target molecule as the root node, it aims to find a complete reaction tree subject to the constraint that all leaf nodes belong to a set of starting materials. The multi-step reactions are crucial because they determine the flow chart in the production of the Organic Chemical Industry. However, existing datasets lack curation of tree-structured multi-step reactions, and fail to provide such reaction trees, limiting models' understanding of organic molecule transformations. In this work, we first develop a benchmark curated for the retrosynthetic planning task, which consists of 124,869 reaction trees retrieved from the public USPTO-full dataset. On top of that, we propose Metro: Memory-Enhanced Transformer for RetrOsynthetic planning. Specifically, the dependency among molecules in the reaction tree is captured as context information for multi-step retrosynthesis predictions through transformers with a memory module. Extensive experiments show that Metro dramatically outperforms existing single-step retrosynthesis models by at least 10.7% in top-1 accuracy. The experiments demonstrate the superiority of exploiting context information in the retrosynthetic planning task. Moreover, the proposed model can be directly used for synthetic accessibility analysis, as it is trained on reaction trees with the shortest depths. Our work is the first step towards a brand new formulation for retrosynthetic planning in the aspects of data construction, model design, and evaluation. Code is available at https://github.com/SongtaoLiu0823/metro.
Debiasing Graph Neural Networks via Learning Disentangled Causal Substructure
Sep 28, 2022
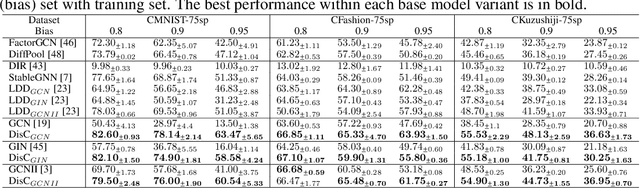
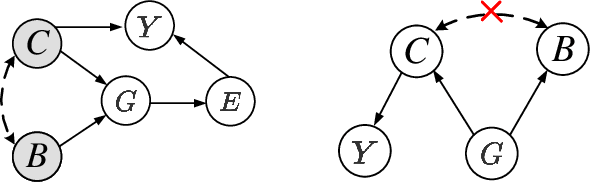
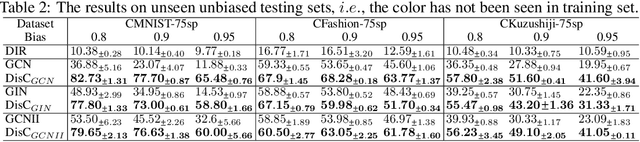
Most Graph Neural Networks (GNNs) predict the labels of unseen graphs by learning the correlation between the input graphs and labels. However, by presenting a graph classification investigation on the training graphs with severe bias, surprisingly, we discover that GNNs always tend to explore the spurious correlations to make decision, even if the causal correlation always exists. This implies that existing GNNs trained on such biased datasets will suffer from poor generalization capability. By analyzing this problem in a causal view, we find that disentangling and decorrelating the causal and bias latent variables from the biased graphs are both crucial for debiasing. Inspiring by this, we propose a general disentangled GNN framework to learn the causal substructure and bias substructure, respectively. Particularly, we design a parameterized edge mask generator to explicitly split the input graph into causal and bias subgraphs. Then two GNN modules supervised by causal/bias-aware loss functions respectively are trained to encode causal and bias subgraphs into their corresponding representations. With the disentangled representations, we synthesize the counterfactual unbiased training samples to further decorrelate causal and bias variables. Moreover, to better benchmark the severe bias problem, we construct three new graph datasets, which have controllable bias degrees and are easier to visualize and explain. Experimental results well demonstrate that our approach achieves superior generalization performance over existing baselines. Furthermore, owing to the learned edge mask, the proposed model has appealing interpretability and transferability. Code and data are available at: https://github.com/googlebaba/DisC.