 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRare Event Analysis via Stochastic Optimal Control
Apr 14, 2026Rare events such as conformational changes in biomolecules, phase transitions, and chemical reactions are central to the behavior of many physical systems, yet they are extremely difficult to study computationally because unbiased simulations seldom produce them. Transition Path Theory (TPT) provides a rigorous statistical framework for analyzing such events: it characterizes the ensemble of reactive trajectories between two designated metastable states (reactant and product), and its central object--the committor function, which gives the probability that the system will next reach the product rather than the reactant--encodes all essential kinetic and thermodynamic information. We introduce a framework that casts committor estimation as a stochastic optimal control (SOC) problem. In this formulation the committor defines a feedback control--proportional to the gradient of its logarithm--that actively steers trajectories toward the reactive region, thereby enabling efficient sampling of reactive paths. To solve the resulting hitting-time control problem we develop two complementary objectives: a direct backpropagation loss and a principled off-policy Value Matching loss, for which we establish first-order optimality guarantees. We further address metastability, which can trap controlled trajectories in intermediate basins, by introducing an alternative sampling process that preserves the reactive current while lowering effective energy barriers. On benchmark systems, the framework yields markedly more accurate committor estimates, reaction rates, and equilibrium constants than existing methods.
A Priori Sampling of Transition States with Guided Diffusion
Mar 26, 2026Transition states, the first-order saddle points on the potential energy surfaces, govern the kinetics and mechanisms of chemical reactions and conformational changes. Locating them is challenging because transition pathways are topologically complex and can proceed via an ensemble of diverse routes. Existing methods address these challenges by introducing heuristic assumptions about the pathway or reaction coordinates, which limits their applicability when a good initial guess is unavailable or when the guess precludes alternative, potentially relevant pathways. We propose to bypass such heuristic limitations by introducing ASTRA, A Priori Sampling of TRAnsition States with Guided Diffusion, which reframes the transition state search as an inference-time scaling problem for generative models. ASTRA trains a score-based diffusion model on configurations from known metastable states. Then, ASTRA guides inference toward the isodensity surface separating the basins of metastable states via a principled composition of conditional scores. A Score-Aligned Ascent (SAA) process then approximates a reaction coordinate from the difference between conditioned scores and combines it with physical forces to drive convergence onto first-order transition states. Validated on benchmarks from 2D potentials to biomolecular conformational changes and chemical reaction, ASTRA locates transition states with high precision and discovers multiple reaction pathways, enabling mechanistic studies of complex molecular systems.
Assessing generative modeling approaches for free energy estimates in condensed matter
Dec 30, 2025The accurate estimation of free energy differences between two states is a long-standing challenge in molecular simulations. Traditional approaches generally rely on sampling multiple intermediate states to ensure sufficient overlap in phase space and are, consequently, computationally expensive. Several generative-model-based methods have recently addressed this challenge by learning a direct bridge between distributions, bypassing the need for intermediate states. However, it remains unclear which approaches provide the best trade-off between efficiency, accuracy, and scalability. In this work, we systematically review these methods and benchmark selected approaches with a focus on condensed-matter systems. In particular, we investigate the performance of discrete and continuous normalizing flows in the context of targeted free energy perturbation as well as FEAT (Free energy Estimators with Adaptive Transport) together with the escorted Jarzynski equality, using coarse-grained monatomic ice and Lennard-Jones solids as benchmark systems. We evaluate accuracy, data efficiency, computational cost, and scalability with system size. Our results provide a quantitative framework for selecting effective free energy estimation strategies in condensed-phase systems.
Trust Region Constrained Measure Transport in Path Space for Stochastic Optimal Control and Inference
Aug 17, 2025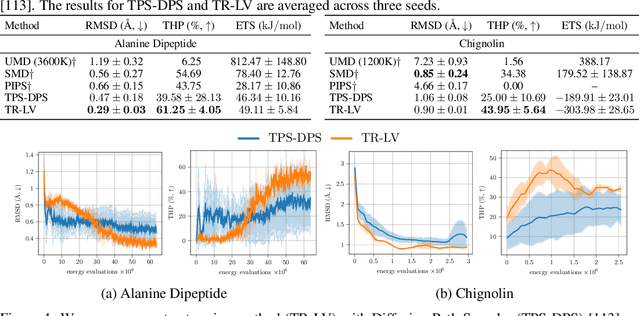



Solving stochastic optimal control problems with quadratic control costs can be viewed as approximating a target path space measure, e.g. via gradient-based optimization. In practice, however, this optimization is challenging in particular if the target measure differs substantially from the prior. In this work, we therefore approach the problem by iteratively solving constrained problems incorporating trust regions that aim for approaching the target measure gradually in a systematic way. It turns out that this trust region based strategy can be understood as a geometric annealing from the prior to the target measure, where, however, the incorporated trust regions lead to a principled and educated way of choosing the time steps in the annealing path. We demonstrate in multiple optimal control applications that our novel method can improve performance significantly, including tasks in diffusion-based sampling, transition path sampling, and fine-tuning of diffusion models.
RNE: a plug-and-play framework for diffusion density estimation and inference-time control
Jun 06, 2025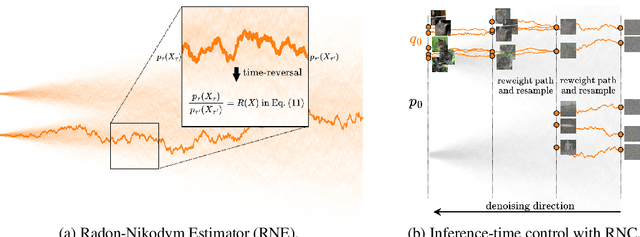
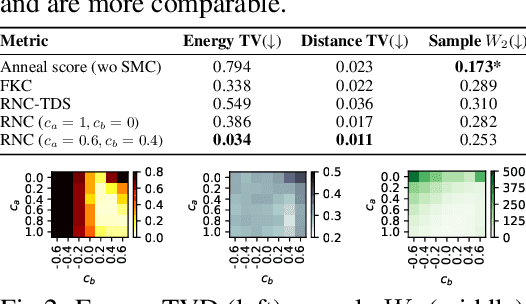

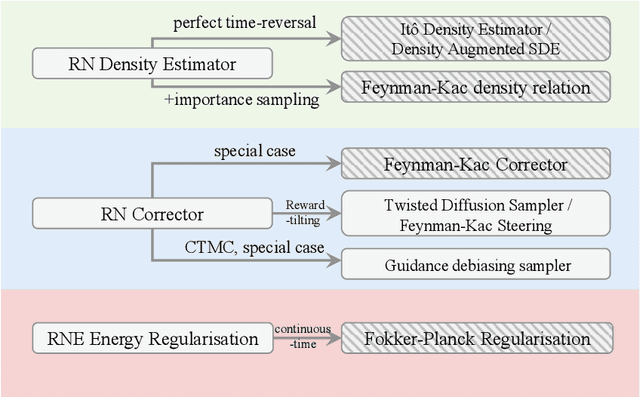
In this paper, we introduce the Radon-Nikodym Estimator (RNE), a flexible, plug-and-play framework for diffusion inference-time density estimation and control, based on the concept of the density ratio between path distributions. RNE connects and unifies a variety of existing density estimation and inference-time control methods under a single and intuitive perspective, stemming from basic variational inference and probabilistic principles therefore offering both theoretical clarity and practical versatility. Experiments demonstrate that RNE achieves promising performances in diffusion density estimation and inference-time control tasks, including annealing, composition of diffusion models, and reward-tilting.
Building-Block Aware Generative Modeling for 3D Crystals of Metal Organic Frameworks
May 13, 2025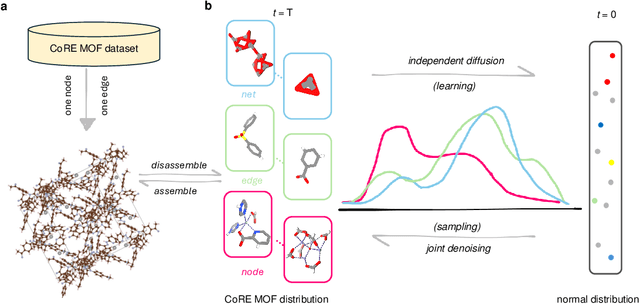
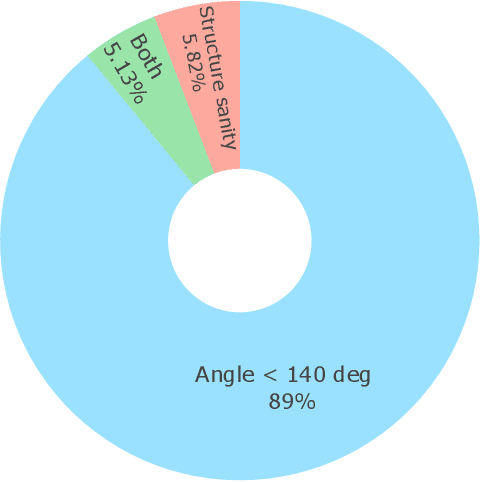
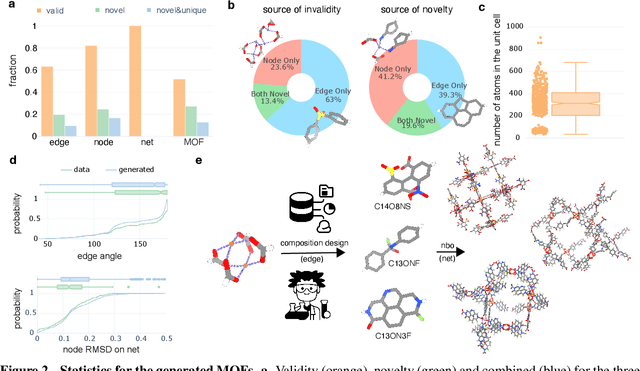
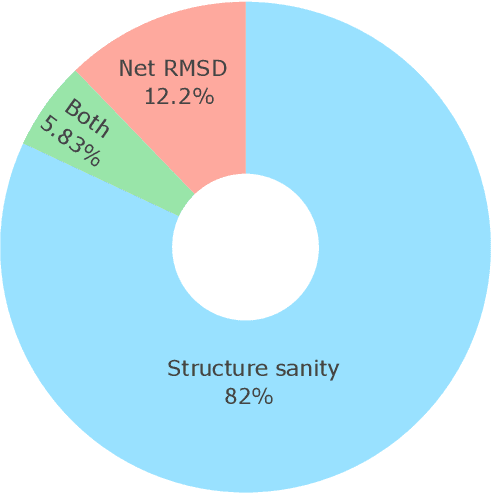
Metal-organic frameworks (MOFs) marry inorganic nodes, organic edges, and topological nets into programmable porous crystals, yet their astronomical design space defies brute-force synthesis. Generative modeling holds ultimate promise, but existing models either recycle known building blocks or are restricted to small unit cells. We introduce Building-Block-Aware MOF Diffusion (BBA MOF Diffusion), an SE(3)-equivariant diffusion model that learns 3D all-atom representations of individual building blocks, encoding crystallographic topological nets explicitly. Trained on the CoRE-MOF database, BBA MOF Diffusion readily samples MOFs with unit cells containing 1000 atoms with great geometric validity, novelty, and diversity mirroring experimental databases. Its native building-block representation produces unprecedented metal nodes and organic edges, expanding accessible chemical space by orders of magnitude. One high-scoring [Zn(1,4-TDC)(EtOH)2] MOF predicted by the model was synthesized, where powder X-ray diffraction, thermogravimetric analysis, and N2 sorption confirm its structural fidelity. BBA-Diff thus furnishes a practical pathway to synthesizable and high-performing MOFs.
LLM-Augmented Chemical Synthesis and Design Decision Programs
May 11, 2025Retrosynthesis, the process of breaking down a target molecule into simpler precursors through a series of valid reactions, stands at the core of organic chemistry and drug development. Although recent machine learning (ML) research has advanced single-step retrosynthetic modeling and subsequent route searches, these solutions remain restricted by the extensive combinatorial space of possible pathways. Concurrently, large language models (LLMs) have exhibited remarkable chemical knowledge, hinting at their potential to tackle complex decision-making tasks in chemistry. In this work, we explore whether LLMs can successfully navigate the highly constrained, multi-step retrosynthesis planning problem. We introduce an efficient scheme for encoding reaction pathways and present a new route-level search strategy, moving beyond the conventional step-by-step reactant prediction. Through comprehensive evaluations, we show that our LLM-augmented approach excels at retrosynthesis planning and extends naturally to the broader challenge of synthesizable molecular design.
FEAT: Free energy Estimators with Adaptive Transport
Apr 15, 2025



We present Free energy Estimators with Adaptive Transport (FEAT), a novel framework for free energy estimation -- a critical challenge across scientific domains. FEAT leverages learned transports implemented via stochastic interpolants and provides consistent, minimum-variance estimators based on escorted Jarzynski equality and controlled Crooks theorem, alongside variational upper and lower bounds on free energy differences. Unifying equilibrium and non-equilibrium methods under a single theoretical framework, FEAT establishes a principled foundation for neural free energy calculations. Experimental validation on toy examples, molecular simulations, and quantum field theory demonstrates improvements over existing learning-based methods.
Large Language Models Are Innate Crystal Structure Generators
Feb 28, 2025



Crystal structure generation is fundamental to materials discovery, enabling the prediction of novel materials with desired properties. While existing approaches leverage Large Language Models (LLMs) through extensive fine-tuning on materials databases, we show that pre-trained LLMs can inherently generate stable crystal structures without additional training. Our novel framework MatLLMSearch integrates pre-trained LLMs with evolutionary search algorithms, achieving a 78.38% metastable rate validated by machine learning interatomic potentials and 31.7% DFT-verified stability via quantum mechanical calculations, outperforming specialized models such as CrystalTextLLM. Beyond crystal structure generation, we further demonstrate that our framework can be readily adapted to diverse materials design tasks, including crystal structure prediction and multi-objective optimization of properties such as deformation energy and bulk modulus, all without fine-tuning. These results establish pre-trained LLMs as versatile and effective tools for materials discovery, opening up new venues for crystal structure generation with reduced computational overhead and broader accessibility.
No Trick, No Treat: Pursuits and Challenges Towards Simulation-free Training of Neural Samplers
Feb 10, 2025



We consider the sampling problem, where the aim is to draw samples from a distribution whose density is known only up to a normalization constant. Recent breakthroughs in generative modeling to approximate a high-dimensional data distribution have sparked significant interest in developing neural network-based methods for this challenging problem. However, neural samplers typically incur heavy computational overhead due to simulating trajectories during training. This motivates the pursuit of simulation-free training procedures of neural samplers. In this work, we propose an elegant modification to previous methods, which allows simulation-free training with the help of a time-dependent normalizing flow. However, it ultimately suffers from severe mode collapse. On closer inspection, we find that nearly all successful neural samplers rely on Langevin preconditioning to avoid mode collapsing. We systematically analyze several popular methods with various objective functions and demonstrate that, in the absence of Langevin preconditioning, most of them fail to adequately cover even a simple target. Finally, we draw attention to a strong baseline by combining the state-of-the-art MCMC method, Parallel Tempering (PT), with an additional generative model to shed light on future explorations of neural samplers.