 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeToken Reduction Should Go Beyond Efficiency in Generative Models -- From Vision, Language to Multimodality
May 23, 2025In Transformer architectures, tokens\textemdash discrete units derived from raw data\textemdash are formed by segmenting inputs into fixed-length chunks. Each token is then mapped to an embedding, enabling parallel attention computations while preserving the input's essential information. Due to the quadratic computational complexity of transformer self-attention mechanisms, token reduction has primarily been used as an efficiency strategy. This is especially true in single vision and language domains, where it helps balance computational costs, memory usage, and inference latency. Despite these advances, this paper argues that token reduction should transcend its traditional efficiency-oriented role in the era of large generative models. Instead, we position it as a fundamental principle in generative modeling, critically influencing both model architecture and broader applications. Specifically, we contend that across vision, language, and multimodal systems, token reduction can: (i) facilitate deeper multimodal integration and alignment, (ii) mitigate "overthinking" and hallucinations, (iii) maintain coherence over long inputs, and (iv) enhance training stability, etc. We reframe token reduction as more than an efficiency measure. By doing so, we outline promising future directions, including algorithm design, reinforcement learning-guided token reduction, token optimization for in-context learning, and broader ML and scientific domains. We highlight its potential to drive new model architectures and learning strategies that improve robustness, increase interpretability, and better align with the objectives of generative modeling.
DD-Ranking: Rethinking the Evaluation of Dataset Distillation
May 19, 2025
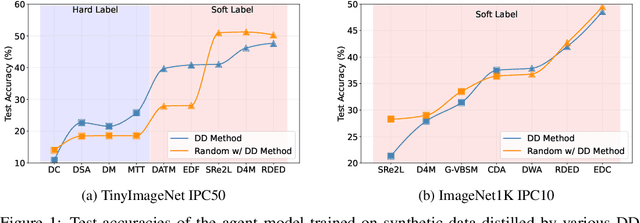
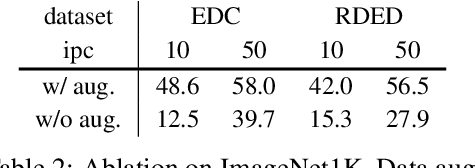
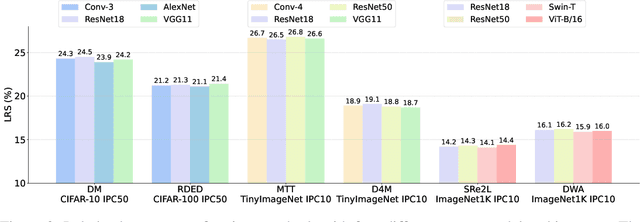
In recent years, dataset distillation has provided a reliable solution for data compression, where models trained on the resulting smaller synthetic datasets achieve performance comparable to those trained on the original datasets. To further improve the performance of synthetic datasets, various training pipelines and optimization objectives have been proposed, greatly advancing the field of dataset distillation. Recent decoupled dataset distillation methods introduce soft labels and stronger data augmentation during the post-evaluation phase and scale dataset distillation up to larger datasets (e.g., ImageNet-1K). However, this raises a question: Is accuracy still a reliable metric to fairly evaluate dataset distillation methods? Our empirical findings suggest that the performance improvements of these methods often stem from additional techniques rather than the inherent quality of the images themselves, with even randomly sampled images achieving superior results. Such misaligned evaluation settings severely hinder the development of DD. Therefore, we propose DD-Ranking, a unified evaluation framework, along with new general evaluation metrics to uncover the true performance improvements achieved by different methods. By refocusing on the actual information enhancement of distilled datasets, DD-Ranking provides a more comprehensive and fair evaluation standard for future research advancements.
ProteinRPN: Towards Accurate Protein Function Prediction with Graph-Based Region Proposals
Sep 01, 2024



Protein function prediction is a crucial task in bioinformatics, with significant implications for understanding biological processes and disease mechanisms. While the relationship between sequence and function has been extensively explored, translating protein structure to function continues to present substantial challenges. Various models, particularly, CNN and graph-based deep learning approaches that integrate structural and functional data, have been proposed to address these challenges. However, these methods often fall short in elucidating the functional significance of key residues essential for protein functionality, as they predominantly adopt a retrospective perspective, leading to suboptimal performance. Inspired by region proposal networks in computer vision, we introduce the Protein Region Proposal Network (ProteinRPN) for accurate protein function prediction. Specifically, the region proposal module component of ProteinRPN identifies potential functional regions (anchors) which are refined through the hierarchy-aware node drop pooling layer favoring nodes with defined secondary structures and spatial proximity. The representations of the predicted functional nodes are enriched using attention mechanisms and subsequently fed into a Graph Multiset Transformer, which is trained with supervised contrastive (SupCon) and InfoNCE losses on perturbed protein structures. Our model demonstrates significant improvements in predicting Gene Ontology (GO) terms, effectively localizing functional residues within protein structures. The proposed framework provides a robust, scalable solution for protein function annotation, advancing the understanding of protein structure-function relationships in computational biology.
TrustLLM: Trustworthiness in Large Language Models
Jan 25, 2024



Large language models (LLMs), exemplified by ChatGPT, have gained considerable attention for their excellent natural language processing capabilities. Nonetheless, these LLMs present many challenges, particularly in the realm of trustworthiness. Therefore, ensuring the trustworthiness of LLMs emerges as an important topic. This paper introduces TrustLLM, a comprehensive study of trustworthiness in LLMs, including principles for different dimensions of trustworthiness, established benchmark, evaluation, and analysis of trustworthiness for mainstream LLMs, and discussion of open challenges and future directions. Specifically, we first propose a set of principles for trustworthy LLMs that span eight different dimensions. Based on these principles, we further establish a benchmark across six dimensions including truthfulness, safety, fairness, robustness, privacy, and machine ethics. We then present a study evaluating 16 mainstream LLMs in TrustLLM, consisting of over 30 datasets. Our findings firstly show that in general trustworthiness and utility (i.e., functional effectiveness) are positively related. Secondly, our observations reveal that proprietary LLMs generally outperform most open-source counterparts in terms of trustworthiness, raising concerns about the potential risks of widely accessible open-source LLMs. However, a few open-source LLMs come very close to proprietary ones. Thirdly, it is important to note that some LLMs may be overly calibrated towards exhibiting trustworthiness, to the extent that they compromise their utility by mistakenly treating benign prompts as harmful and consequently not responding. Finally, we emphasize the importance of ensuring transparency not only in the models themselves but also in the technologies that underpin trustworthiness. Knowing the specific trustworthy technologies that have been employed is crucial for analyzing their effectiveness.
Contextualized Machine Learning
Oct 17, 2023



We examine Contextualized Machine Learning (ML), a paradigm for learning heterogeneous and context-dependent effects. Contextualized ML estimates heterogeneous functions by applying deep learning to the meta-relationship between contextual information and context-specific parametric models. This is a form of varying-coefficient modeling that unifies existing frameworks including cluster analysis and cohort modeling by introducing two reusable concepts: a context encoder which translates sample context into model parameters, and sample-specific model which operates on sample predictors. We review the process of developing contextualized models, nonparametric inference from contextualized models, and identifiability conditions of contextualized models. Finally, we present the open-source PyTorch package ContextualizedML.
LLMs Understand Glass-Box Models, Discover Surprises, and Suggest Repairs
Aug 07, 2023We show that large language models (LLMs) are remarkably good at working with interpretable models that decompose complex outcomes into univariate graph-represented components. By adopting a hierarchical approach to reasoning, LLMs can provide comprehensive model-level summaries without ever requiring the entire model to fit in context. This approach enables LLMs to apply their extensive background knowledge to automate common tasks in data science such as detecting anomalies that contradict prior knowledge, describing potential reasons for the anomalies, and suggesting repairs that would remove the anomalies. We use multiple examples in healthcare to demonstrate the utility of these new capabilities of LLMs, with particular emphasis on Generalized Additive Models (GAMs). Finally, we present the package $\texttt{TalkToEBM}$ as an open-source LLM-GAM interface.
NOTMAD: Estimating Bayesian Networks with Sample-Specific Structures and Parameters
Nov 01, 2021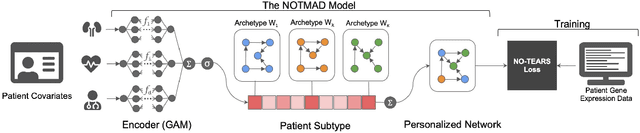


Context-specific Bayesian networks (i.e. directed acyclic graphs, DAGs) identify context-dependent relationships between variables, but the non-convexity induced by the acyclicity requirement makes it difficult to share information between context-specific estimators (e.g. with graph generator functions). For this reason, existing methods for inferring context-specific Bayesian networks have favored breaking datasets into subsamples, limiting statistical power and resolution, and preventing the use of multidimensional and latent contexts. To overcome this challenge, we propose NOTEARS-optimized Mixtures of Archetypal DAGs (NOTMAD). NOTMAD models context-specific Bayesian networks as the output of a function which learns to mix archetypal networks according to sample context. The archetypal networks are estimated jointly with the context-specific networks and do not require any prior knowledge. We encode the acyclicity constraint as a smooth regularization loss which is back-propagated to the mixing function; in this way, NOTMAD shares information between context-specific acyclic graphs, enabling the estimation of Bayesian network structures and parameters at even single-sample resolution. We demonstrate the utility of NOTMAD and sample-specific network inference through analysis and experiments, including patient-specific gene expression networks which correspond to morphological variation in cancer.
Causal Mediation Analysis Leveraging Multiple Types of Summary Statistics Data
Jan 24, 2019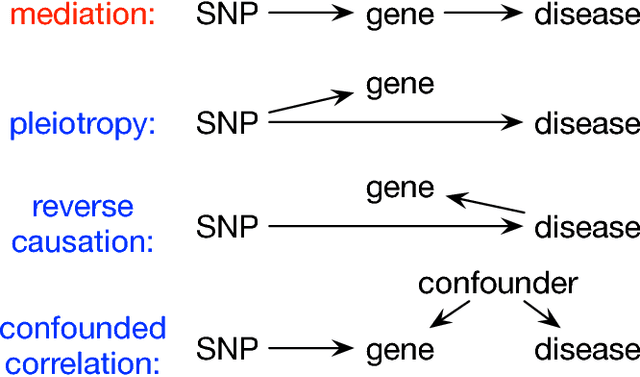
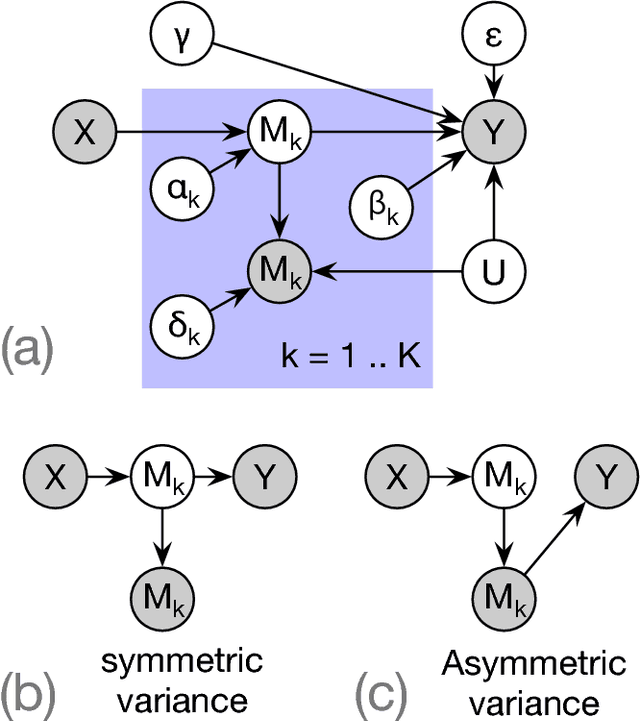
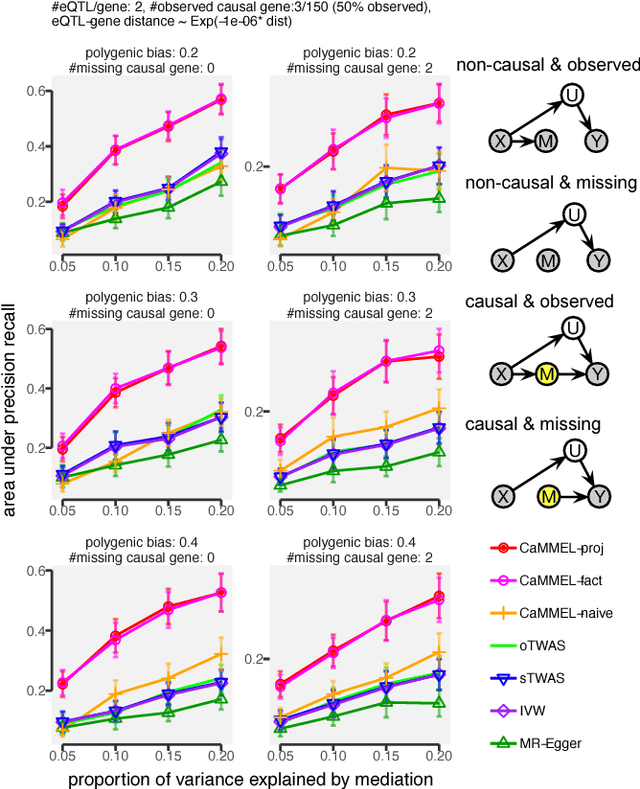
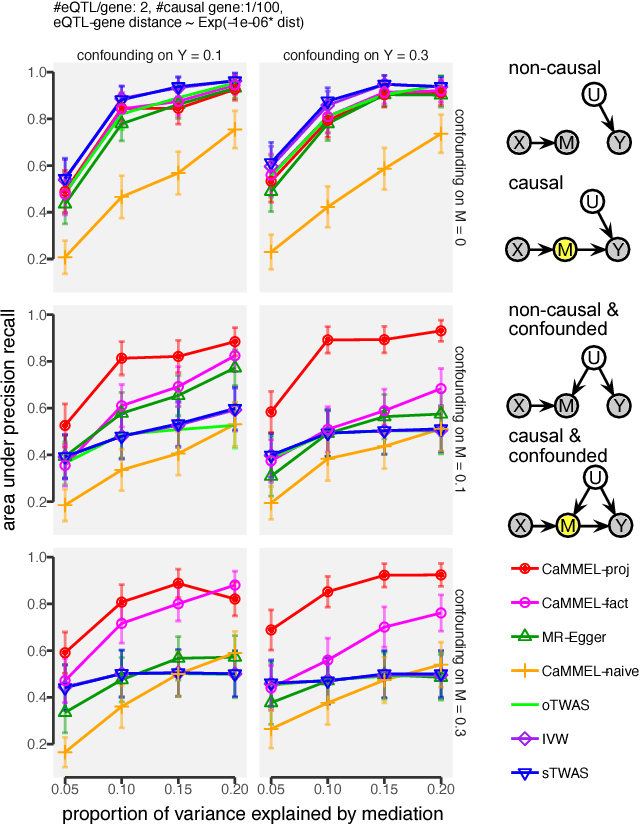
Summary statistics of genome-wide association studies (GWAS) teach causal relationship between millions of genetic markers and tens and thousands of phenotypes. However, underlying biological mechanisms are yet to be elucidated. We can achieve necessary interpretation of GWAS in a causal mediation framework, looking to establish a sparse set of mediators between genetic and downstream variables, but there are several challenges. Unlike existing methods rely on strong and unrealistic assumptions, we tackle practical challenges within a principled summary-based causal inference framework. We analyzed the proposed methods in extensive simulations generated from real-world genetic data. We demonstrated only our approach can accurately redeem causal genes, even without knowing actual individual-level data, despite the presence of competing non-causal trails.
A latent topic model for mining heterogenous non-randomly missing electronic health records data
Nov 01, 2018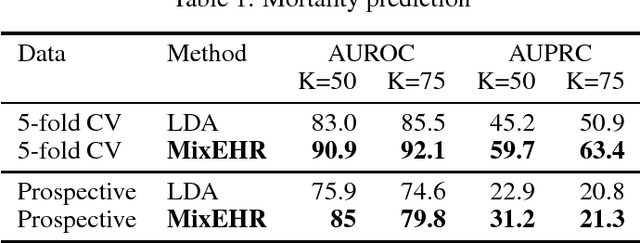
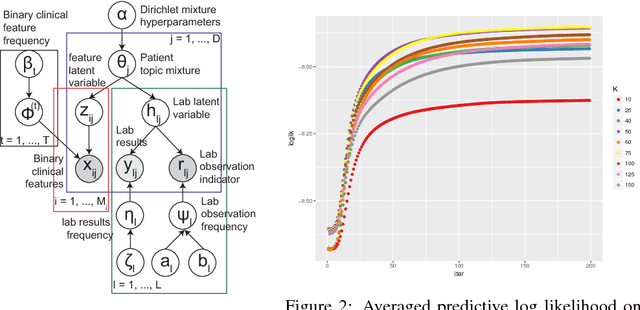
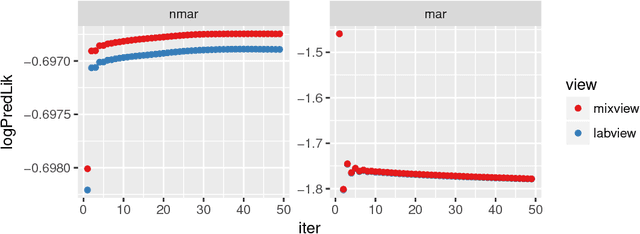
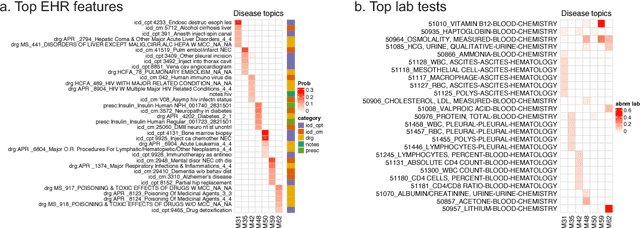
Electronic health records (EHR) are rich heterogeneous collection of patient health information, whose broad adoption provides great opportunities for systematic health data mining. However, heterogeneous EHR data types and biased ascertainment impose computational challenges. Here, we present mixEHR, an unsupervised generative model integrating collaborative filtering and latent topic models, which jointly models the discrete distributions of data observation bias and actual data using latent disease-topic distributions. We apply mixEHR on 12.8 million phenotypic observations from the MIMIC dataset, and use it to reveal latent disease topics, interpret EHR results, impute missing data, and predict mortality in intensive care units. Using both simulation and real data, we show that mixEHR outperforms previous methods and reveals meaningful multi-disease insights.
Network Maximal Correlation
Feb 09, 2017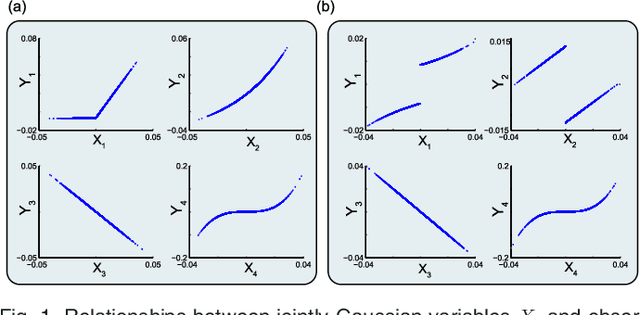
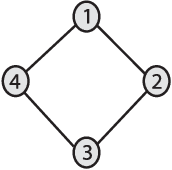
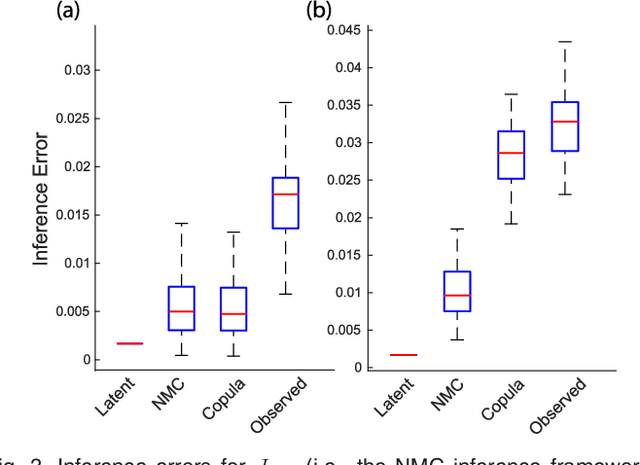
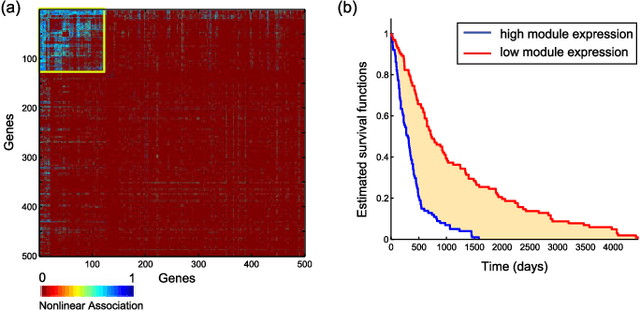
We introduce Network Maximal Correlation (NMC) as a multivariate measure of nonlinear association among random variables. NMC is defined via an optimization that infers transformations of variables by maximizing aggregate inner products between transformed variables. For finite discrete and jointly Gaussian random variables, we characterize a solution of the NMC optimization using basis expansion of functions over appropriate basis functions. For finite discrete variables, we propose an algorithm based on alternating conditional expectation to determine NMC. Moreover we propose a distributed algorithm to compute an approximation of NMC for large and dense graphs using graph partitioning. For finite discrete variables, we show that the probability of discrepancy greater than any given level between NMC and NMC computed using empirical distributions decays exponentially fast as the sample size grows. For jointly Gaussian variables, we show that under some conditions the NMC optimization is an instance of the Max-Cut problem. We then illustrate an application of NMC in inference of graphical model for bijective functions of jointly Gaussian variables. Finally, we show NMC's utility in a data application of learning nonlinear dependencies among genes in a cancer dataset.