 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCausalGaze: Unveiling Hallucinations via Counterfactual Graph Intervention in Large Language Models
Apr 13, 2026Despite the groundbreaking advancements made by large language models (LLMs), hallucination remains a critical bottleneck for their deployment in high-stakes domains. Existing classification-based methods mainly rely on static and passive signals from internal states, which often captures the noise and spurious correlations, while overlooking the underlying causal mechanisms. To address this limitation, we shift the paradigm from passive observation to active intervention by introducing CausalGaze, a novel hallucination detection framework based on structural causal models (SCMs). CausalGaze models LLMs' internal states as dynamic causal graphs and employs counterfactual interventions to disentangle causal reasoning paths from incidental noise, thereby enhancing model interpretability. Extensive experiments across four datasets and three widely used LLMs demonstrate the effectiveness of CausalGaze, especially achieving over 5.2\% improvement in AUROC on the TruthfulQA dataset compared to state-of-the-art baselines.
How Independent are Large Language Models? A Statistical Framework for Auditing Behavioral Entanglement and Reweighting Verifier Ensembles
Apr 08, 2026The rapid growth of the large language model (LLM) ecosystem raises a critical question: are seemingly diverse models truly independent? Shared pretraining data, distillation, and alignment pipelines can induce hidden behavioral dependencies, latent entanglement, that undermine multi-model systems such as LLM-as-a-judge pipelines and ensemble verification, which implicitly assume independent signals. In practice, this manifests as correlated reasoning patterns and synchronized failures, where apparent agreement reflects shared error modes rather than independent validation. To address this, we develop a statistical framework for auditing behavioral entanglement among black-box LLMs. Our approach introduces a multi-resolution hierarchy that characterizes the joint failure manifold through two information-theoretic metrics: (i) a Difficulty-Weighted Behavioral Entanglement Index, which amplifies synchronized failures on easy tasks, and (ii) a Cumulative Information Gain (CIG) metric, which captures directional alignment in erroneous responses. Through extensive experiments on 18 LLMs from six model families, we identify widespread behavioral entanglement and analyze its impact on LLM-as-a-judge evaluation. We find that CIG exhibits a statistically significant association with degradation in judge precision, with Spearman coefficient of 0.64 (p < 0.001) for GPT-4o-mini and 0.71 (p < 0.01) for Llama3-based judges, indicating that stronger dependency corresponds to increased over-endorsement bias. Finally, we demonstrate a practical use case of entanglement through de-entangled verifier ensemble reweighting. By adjusting model contributions based on inferred independence, the proposed method mitigates correlated bias and improves verification performance, achieving up to a 4.5% accuracy gain over majority voting.
Knowledge-Grounded Agentic Large Language Models for Multi-Hazard Understanding from Reconnaissance Reports
Nov 19, 2025Post-disaster reconnaissance reports contain critical evidence for understanding multi-hazard interactions, yet their unstructured narratives make systematic knowledge transfer difficult. Large language models (LLMs) offer new potential for analyzing these reports, but often generate unreliable or hallucinated outputs when domain grounding is absent. This study introduces the Mixture-of-Retrieval Agentic RAG (MoRA-RAG), a knowledge-grounded LLM framework that transforms reconnaissance reports into a structured foundation for multi-hazard reasoning. The framework integrates a Mixture-of-Retrieval mechanism that dynamically routes queries across hazard-specific databases while using agentic chunking to preserve contextual coherence during retrieval. It also includes a verification loop that assesses evidence sufficiency, refines queries, and initiates targeted searches when information remains incomplete. We construct HazardRecQA by deriving question-answer pairs from GEER reconnaissance reports, which document 90 global events across seven major hazard types. MoRA-RAG achieves up to 94.5 percent accuracy, outperforming zero-shot LLMs by 30 percent and state-of-the-art RAG systems by 10 percent, while reducing hallucinations across diverse LLM architectures. MoRA-RAG also enables open-weight LLMs to achieve performance comparable to proprietary models. It establishes a new paradigm for transforming post-disaster documentation into actionable, trustworthy intelligence for hazard resilience.
Simulating the Unseen: Crash Prediction Must Learn from What Did Not Happen
May 27, 2025Traffic safety science has long been hindered by a fundamental data paradox: the crashes we most wish to prevent are precisely those events we rarely observe. Existing crash-frequency models and surrogate safety metrics rely heavily on sparse, noisy, and under-reported records, while even sophisticated, high-fidelity simulations undersample the long-tailed situations that trigger catastrophic outcomes such as fatalities. We argue that the path to achieving Vision Zero, i.e., the complete elimination of traffic fatalities and severe injuries, requires a paradigm shift from traditional crash-only learning to a new form of counterfactual safety learning: reasoning not only about what happened, but also about the vast set of plausible yet perilous scenarios that could have happened under slightly different circumstances. To operationalize this shift, our proposed agenda bridges macro to micro. Guided by crash-rate priors, generative scene engines, diverse driver models, and causal learning, near-miss events are synthesized and explained. A crash-focused digital twin testbed links micro scenes to macro patterns, while a multi-objective validator ensures that simulations maintain statistical realism. This pipeline transforms sparse crash data into rich signals for crash prediction, enabling the stress-testing of vehicles, roads, and policies before deployment. By learning from crashes that almost happened, we can shift traffic safety from reactive forensics to proactive prevention, advancing Vision Zero.
NuScenes-SpatialQA: A Spatial Understanding and Reasoning Benchmark for Vision-Language Models in Autonomous Driving
Apr 07, 2025



Recent advancements in Vision-Language Models (VLMs) have demonstrated strong potential for autonomous driving tasks. However, their spatial understanding and reasoning-key capabilities for autonomous driving-still exhibit significant limitations. Notably, none of the existing benchmarks systematically evaluate VLMs' spatial reasoning capabilities in driving scenarios. To fill this gap, we propose NuScenes-SpatialQA, the first large-scale ground-truth-based Question-Answer (QA) benchmark specifically designed to evaluate the spatial understanding and reasoning capabilities of VLMs in autonomous driving. Built upon the NuScenes dataset, the benchmark is constructed through an automated 3D scene graph generation pipeline and a QA generation pipeline. The benchmark systematically evaluates VLMs' performance in both spatial understanding and reasoning across multiple dimensions. Using this benchmark, we conduct extensive experiments on diverse VLMs, including both general and spatial-enhanced models, providing the first comprehensive evaluation of their spatial capabilities in autonomous driving. Surprisingly, the experimental results show that the spatial-enhanced VLM outperforms in qualitative QA but does not demonstrate competitiveness in quantitative QA. In general, VLMs still face considerable challenges in spatial understanding and reasoning.
PathVQ: Reforming Computational Pathology Foundation Model for Whole Slide Image Analysis via Vector Quantization
Mar 09, 2025Computational pathology and whole-slide image (WSI) analysis are pivotal in cancer diagnosis and prognosis. However, the ultra-high resolution of WSIs presents significant modeling challenges. Recent advancements in pathology foundation models have improved performance, yet most approaches rely on [CLS] token representation of tile ViT as slide-level inputs (16x16 pixels is refereed as patch and 224x224 pixels as tile). This discards critical spatial details from patch tokens, limiting downstream WSI analysis tasks. We find that leveraging all spatial patch tokens benefits WSI analysis but incurs nearly 200x higher storage and training costs (e.g., 196 tokens in ViT$_{224}$). To address this, we introduce vector quantized (VQ) distillation on patch feature, which efficiently compresses spatial patch tokens using discrete indices and a decoder. Our method reduces token dimensionality from 1024 to 16, achieving a 64x compression rate while preserving reconstruction fidelity. Furthermore, we employ a multi-scale VQ (MSVQ) strategy, which not only enhances VQ reconstruction performance but also serves as a Self-supervised Learning (SSL) supervision for a seamless slide-level pretraining objective. Built upon the quantized patch features and supervision targets of tile via MSVQ, we develop a progressive convolutional module and slide-level SSL to extract representations with rich spatial-information for downstream WSI tasks. Extensive evaluations on multiple datasets demonstrate the effectiveness of our approach, achieving state-of-the-art performance in WSI analysis. Code will be available soon.
Towards Effective and Efficient Context-aware Nucleus Detection in Histopathology Whole Slide Images
Mar 04, 2025
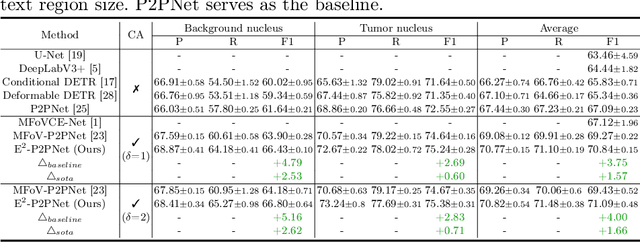
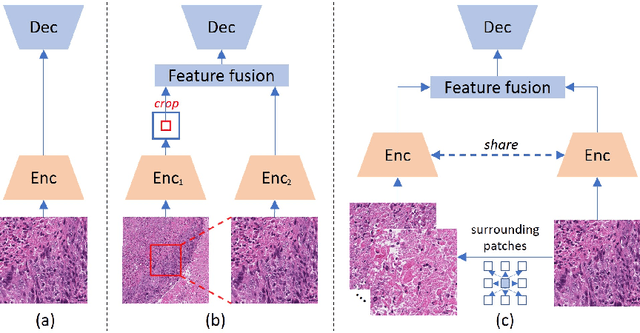
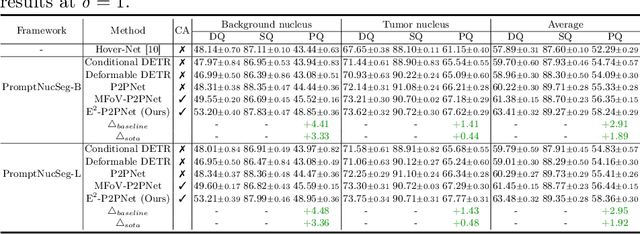
Nucleus detection in histopathology whole slide images (WSIs) is crucial for a broad spectrum of clinical applications. Current approaches for nucleus detection in gigapixel WSIs utilize a sliding window methodology, which overlooks boarder contextual information (eg, tissue structure) and easily leads to inaccurate predictions. To address this problem, recent studies additionally crops a large Filed-of-View (FoV) region around each sliding window to extract contextual features. However, such methods substantially increases the inference latency. In this paper, we propose an effective and efficient context-aware nucleus detection algorithm. Specifically, instead of leveraging large FoV regions, we aggregate contextual clues from off-the-shelf features of historically visited sliding windows. This design greatly reduces computational overhead. Moreover, compared to large FoV regions at a low magnification, the sliding window patches have higher magnification and provide finer-grained tissue details, thereby enhancing the detection accuracy. To further improve the efficiency, we propose a grid pooling technique to compress dense feature maps of each patch into a few contextual tokens. Finally, we craft OCELOT-seg, the first benchmark dedicated to context-aware nucleus instance segmentation. Code, dataset, and model checkpoints will be available at https://github.com/windygoo/PathContext.
Rethinking Transformer for Long Contextual Histopathology Whole Slide Image Analysis
Oct 18, 2024



Histopathology Whole Slide Image (WSI) analysis serves as the gold standard for clinical cancer diagnosis in the daily routines of doctors. To develop computer-aided diagnosis model for WSIs, previous methods typically employ Multi-Instance Learning to enable slide-level prediction given only slide-level labels. Among these models, vanilla attention mechanisms without pairwise interactions have traditionally been employed but are unable to model contextual information. More recently, self-attention models have been utilized to address this issue. To alleviate the computational complexity of long sequences in large WSIs, methods like HIPT use region-slicing, and TransMIL employs approximation of full self-attention. Both approaches suffer from suboptimal performance due to the loss of key information. Moreover, their use of absolute positional embedding struggles to effectively handle long contextual dependencies in shape-varying WSIs. In this paper, we first analyze how the low-rank nature of the long-sequence attention matrix constrains the representation ability of WSI modelling. Then, we demonstrate that the rank of attention matrix can be improved by focusing on local interactions via a local attention mask. Our analysis shows that the local mask aligns with the attention patterns in the lower layers of the Transformer. Furthermore, the local attention mask can be implemented during chunked attention calculation, reducing the quadratic computational complexity to linear with a small local bandwidth. Building on this, we propose a local-global hybrid Transformer for both computational acceleration and local-global information interactions modelling. Our method, Long-contextual MIL (LongMIL), is evaluated through extensive experiments on various WSI tasks to validate its superiority. Our code will be available at github.com/invoker-LL/Long-MIL.
Large-scale cervical precancerous screening via AI-assisted cytology whole slide image analysis
Jul 28, 2024



Cervical Cancer continues to be the leading gynecological malignancy, posing a persistent threat to women's health on a global scale. Early screening via cytology Whole Slide Image (WSI) diagnosis is critical to prevent this Cancer progression and improve survival rate, but pathologist's single test suffers inevitable false negative due to the immense number of cells that need to be reviewed within a WSI. Though computer-aided automated diagnostic models can serve as strong complement for pathologists, their effectiveness is hampered by the paucity of extensive and detailed annotations, coupled with the limited interpretability and robustness. These factors significantly hinder their practical applicability and reliability in clinical settings. To tackle these challenges, we develop an AI approach, which is a Scalable Technology for Robust and Interpretable Diagnosis built on Extensive data (STRIDE) of cervical cytology. STRIDE addresses the bottleneck of limited annotations by integrating patient-level labels with a small portion of cell-level labels through an end-to-end training strategy, facilitating scalable learning across extensive datasets. To further improve the robustness to real-world domain shifts of cytology slide-making and imaging, STRIDE employs color adversarial samples training that mimic staining and imaging variations. Lastly, to achieve pathologist-level interpretability for the trustworthiness in clinical settings, STRIDE can generate explanatory textual descriptions that simulates pathologists' diagnostic processes by cell image feature and textual description alignment. Conducting extensive experiments and evaluations in 183 medical centers with a dataset of 341,889 WSIs and 0.1 billion cells from cervical cytology patients, STRIDE has demonstrated a remarkable superiority over previous state-of-the-art techniques.
PathGen-1.6M: 1.6 Million Pathology Image-text Pairs Generation through Multi-agent Collaboration
Jun 28, 2024



Vision Language Models (VLMs) like CLIP have attracted substantial attention in pathology, serving as backbones for applications such as zero-shot image classification and Whole Slide Image (WSI) analysis. Additionally, they can function as vision encoders when combined with large language models (LLMs) to support broader capabilities. Current efforts to train pathology VLMs rely on pathology image-text pairs from platforms like PubMed, YouTube, and Twitter, which provide limited, unscalable data with generally suboptimal image quality. In this work, we leverage large-scale WSI datasets like TCGA to extract numerous high-quality image patches. We then train a large multimodal model to generate captions for these images, creating PathGen-1.6M, a dataset containing 1.6 million high-quality image-caption pairs. Our approach involves multiple agent models collaborating to extract representative WSI patches, generating and refining captions to obtain high-quality image-text pairs. Extensive experiments show that integrating these generated pairs with existing datasets to train a pathology-specific CLIP model, PathGen-CLIP, significantly enhances its ability to analyze pathological images, with substantial improvements across nine pathology-related zero-shot image classification tasks and three whole-slide image tasks. Furthermore, we construct 200K instruction-tuning data based on PathGen-1.6M and integrate PathGen-CLIP with the Vicuna LLM to create more powerful multimodal models through instruction tuning. Overall, we provide a scalable pathway for high-quality data generation in pathology, paving the way for next-generation general pathology models.