 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCodeDance: A Dynamic Tool-integrated MLLM for Executable Visual Reasoning
Dec 19, 2025Recent releases such as o3 highlight human-like "thinking with images" reasoning that combines structured tool use with stepwise verification, yet most open-source approaches still rely on text-only chains, rigid visual schemas, or single-step pipelines, limiting flexibility, interpretability, and transferability on complex tasks. We introduce CodeDance, which explores executable code as a general solver for visual reasoning. Unlike fixed-schema calls (e.g., only predicting bounding-box coordinates), CodeDance defines, composes, and executes code to orchestrate multiple tools, compute intermediate results, and render visual artifacts (e.g., boxes, lines, plots) that support transparent, self-checkable reasoning. To guide this process, we introduce a reward for balanced and adaptive tool-call, which balances exploration with efficiency and mitigates tool overuse. Interestingly, beyond the expected capabilities taught by atomic supervision, we empirically observe novel emergent behaviors during RL training: CodeDance demonstrates novel tool invocations, unseen compositions, and cross-task transfer. These behaviors arise without task-specific fine-tuning, suggesting a general and scalable mechanism of executable visual reasoning. Extensive experiments across reasoning benchmarks (e.g., visual search, math, chart QA) show that CodeDance not only consistently outperforms schema-driven and text-only baselines, but also surpasses advanced closed models such as GPT-4o and larger open-source models.
SAC-MIL: Spatial-Aware Correlated Multiple Instance Learning for Histopathology Whole Slide Image Classification
Sep 04, 2025



We propose Spatial-Aware Correlated Multiple Instance Learning (SAC-MIL) for performing WSI classification. SAC-MIL consists of a positional encoding module to encode position information and a SAC block to perform full instance correlations. The positional encoding module utilizes the instance coordinates within the slide to encode the spatial relationships instead of the instance index in the input WSI sequence. The positional encoding module can also handle the length extrapolation issue where the training and testing sequences have different lengths. The SAC block is an MLP-based method that performs full instance correlation in linear time complexity with respect to the sequence length. Due to the simple structure of MLP, it is easy to deploy since it does not require custom CUDA kernels, compared to Transformer-based methods for WSI classification. SAC-MIL has achieved state-of-the-art performance on the CAMELYON-16, TCGA-LUNG, and TCGA-BRAC datasets. The code will be released upon acceptance.
PathVQ: Reforming Computational Pathology Foundation Model for Whole Slide Image Analysis via Vector Quantization
Mar 09, 2025Computational pathology and whole-slide image (WSI) analysis are pivotal in cancer diagnosis and prognosis. However, the ultra-high resolution of WSIs presents significant modeling challenges. Recent advancements in pathology foundation models have improved performance, yet most approaches rely on [CLS] token representation of tile ViT as slide-level inputs (16x16 pixels is refereed as patch and 224x224 pixels as tile). This discards critical spatial details from patch tokens, limiting downstream WSI analysis tasks. We find that leveraging all spatial patch tokens benefits WSI analysis but incurs nearly 200x higher storage and training costs (e.g., 196 tokens in ViT$_{224}$). To address this, we introduce vector quantized (VQ) distillation on patch feature, which efficiently compresses spatial patch tokens using discrete indices and a decoder. Our method reduces token dimensionality from 1024 to 16, achieving a 64x compression rate while preserving reconstruction fidelity. Furthermore, we employ a multi-scale VQ (MSVQ) strategy, which not only enhances VQ reconstruction performance but also serves as a Self-supervised Learning (SSL) supervision for a seamless slide-level pretraining objective. Built upon the quantized patch features and supervision targets of tile via MSVQ, we develop a progressive convolutional module and slide-level SSL to extract representations with rich spatial-information for downstream WSI tasks. Extensive evaluations on multiple datasets demonstrate the effectiveness of our approach, achieving state-of-the-art performance in WSI analysis. Code will be available soon.
Towards Effective and Efficient Context-aware Nucleus Detection in Histopathology Whole Slide Images
Mar 04, 2025
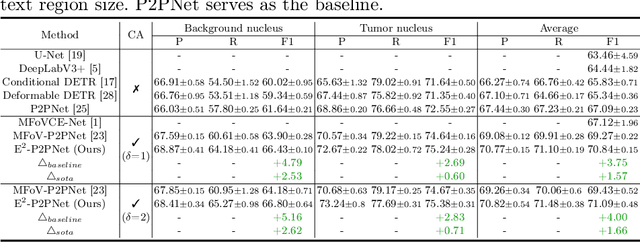
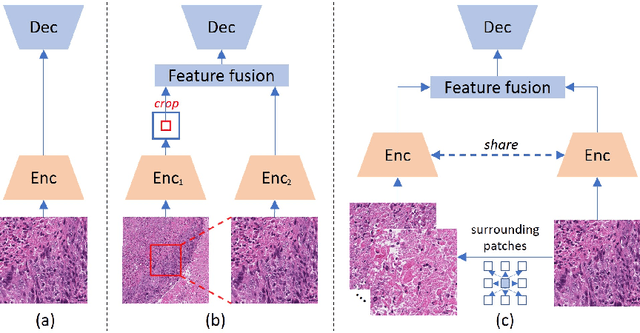
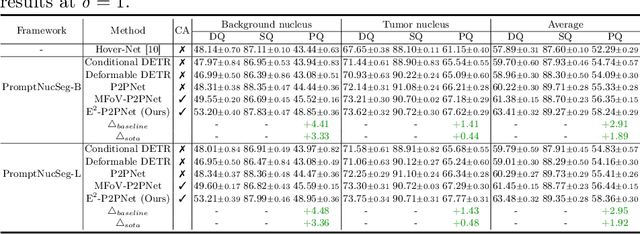
Nucleus detection in histopathology whole slide images (WSIs) is crucial for a broad spectrum of clinical applications. Current approaches for nucleus detection in gigapixel WSIs utilize a sliding window methodology, which overlooks boarder contextual information (eg, tissue structure) and easily leads to inaccurate predictions. To address this problem, recent studies additionally crops a large Filed-of-View (FoV) region around each sliding window to extract contextual features. However, such methods substantially increases the inference latency. In this paper, we propose an effective and efficient context-aware nucleus detection algorithm. Specifically, instead of leveraging large FoV regions, we aggregate contextual clues from off-the-shelf features of historically visited sliding windows. This design greatly reduces computational overhead. Moreover, compared to large FoV regions at a low magnification, the sliding window patches have higher magnification and provide finer-grained tissue details, thereby enhancing the detection accuracy. To further improve the efficiency, we propose a grid pooling technique to compress dense feature maps of each patch into a few contextual tokens. Finally, we craft OCELOT-seg, the first benchmark dedicated to context-aware nucleus instance segmentation. Code, dataset, and model checkpoints will be available at https://github.com/windygoo/PathContext.
Panther: Illuminate the Sight of Multimodal LLMs with Instruction-Guided Visual Prompts
Nov 22, 2024



Multimodal large language models (MLLMs) are closing the gap to human visual perception capability rapidly, while, still lag behind on attending to subtle images details or locating small objects precisely, etc. Common schemes to tackle these issues include deploying multiple vision encoders or operating on original high-resolution images. Few studies have concentrated on taking the textual instruction into improving visual representation, resulting in losing focus in some vision-centric tasks, a phenomenon we herein termed as Amblyopia. In this work, we introduce Panther, a MLLM that closely adheres to user instruction and locates targets of interests precisely, with the finesse of a black panther. Specifically, Panther comprises three integral components: Panther-VE, Panther-Bridge, and Panther-Decoder. Panther-VE integrates user instruction information at the early stages of the vision encoder, thereby extracting the most relevant and useful visual representations. The Panther-Bridge module, equipped with powerful filtering capabilities, significantly reduces redundant visual information, leading to a substantial savings in training costs. The Panther-Decoder is versatile and can be employed with any decoder-only architecture of LLMs without discrimination. Experimental results, particularly on vision-centric benchmarks, have demonstrated the effectiveness of Panther.
Rethinking Transformer for Long Contextual Histopathology Whole Slide Image Analysis
Oct 18, 2024



Histopathology Whole Slide Image (WSI) analysis serves as the gold standard for clinical cancer diagnosis in the daily routines of doctors. To develop computer-aided diagnosis model for WSIs, previous methods typically employ Multi-Instance Learning to enable slide-level prediction given only slide-level labels. Among these models, vanilla attention mechanisms without pairwise interactions have traditionally been employed but are unable to model contextual information. More recently, self-attention models have been utilized to address this issue. To alleviate the computational complexity of long sequences in large WSIs, methods like HIPT use region-slicing, and TransMIL employs approximation of full self-attention. Both approaches suffer from suboptimal performance due to the loss of key information. Moreover, their use of absolute positional embedding struggles to effectively handle long contextual dependencies in shape-varying WSIs. In this paper, we first analyze how the low-rank nature of the long-sequence attention matrix constrains the representation ability of WSI modelling. Then, we demonstrate that the rank of attention matrix can be improved by focusing on local interactions via a local attention mask. Our analysis shows that the local mask aligns with the attention patterns in the lower layers of the Transformer. Furthermore, the local attention mask can be implemented during chunked attention calculation, reducing the quadratic computational complexity to linear with a small local bandwidth. Building on this, we propose a local-global hybrid Transformer for both computational acceleration and local-global information interactions modelling. Our method, Long-contextual MIL (LongMIL), is evaluated through extensive experiments on various WSI tasks to validate its superiority. Our code will be available at github.com/invoker-LL/Long-MIL.
Large-scale cervical precancerous screening via AI-assisted cytology whole slide image analysis
Jul 28, 2024



Cervical Cancer continues to be the leading gynecological malignancy, posing a persistent threat to women's health on a global scale. Early screening via cytology Whole Slide Image (WSI) diagnosis is critical to prevent this Cancer progression and improve survival rate, but pathologist's single test suffers inevitable false negative due to the immense number of cells that need to be reviewed within a WSI. Though computer-aided automated diagnostic models can serve as strong complement for pathologists, their effectiveness is hampered by the paucity of extensive and detailed annotations, coupled with the limited interpretability and robustness. These factors significantly hinder their practical applicability and reliability in clinical settings. To tackle these challenges, we develop an AI approach, which is a Scalable Technology for Robust and Interpretable Diagnosis built on Extensive data (STRIDE) of cervical cytology. STRIDE addresses the bottleneck of limited annotations by integrating patient-level labels with a small portion of cell-level labels through an end-to-end training strategy, facilitating scalable learning across extensive datasets. To further improve the robustness to real-world domain shifts of cytology slide-making and imaging, STRIDE employs color adversarial samples training that mimic staining and imaging variations. Lastly, to achieve pathologist-level interpretability for the trustworthiness in clinical settings, STRIDE can generate explanatory textual descriptions that simulates pathologists' diagnostic processes by cell image feature and textual description alignment. Conducting extensive experiments and evaluations in 183 medical centers with a dataset of 341,889 WSIs and 0.1 billion cells from cervical cytology patients, STRIDE has demonstrated a remarkable superiority over previous state-of-the-art techniques.
WSI-VQA: Interpreting Whole Slide Images by Generative Visual Question Answering
Jul 08, 2024



Whole slide imaging is routinely adopted for carcinoma diagnosis and prognosis. Abundant experience is required for pathologists to achieve accurate and reliable diagnostic results of whole slide images (WSI). The huge size and heterogeneous features of WSIs make the workflow of pathological reading extremely time-consuming. In this paper, we propose a novel framework (WSI-VQA) to interpret WSIs by generative visual question answering. WSI-VQA shows universality by reframing various kinds of slide-level tasks in a question-answering pattern, in which pathologists can achieve immunohistochemical grading, survival prediction, and tumor subtyping following human-machine interaction. Furthermore, we establish a WSI-VQA dataset which contains 8672 slide-level question-answering pairs with 977 WSIs. Besides the ability to deal with different slide-level tasks, our generative model which is named Wsi2Text Transformer (W2T) outperforms existing discriminative models in medical correctness, which reveals the potential of our model to be applied in the clinical scenario. Additionally, we also visualize the co-attention mapping between word embeddings and WSIs as an intuitive explanation for diagnostic results. The dataset and related code are available at https://github.com/cpystan/WSI-VQA.
ADR: Attention Diversification Regularization for Mitigating Overfitting in Multiple Instance Learning based Whole Slide Image Classification
Jun 18, 2024



Multiple Instance Learning (MIL) has demonstrated effectiveness in analyzing whole slide images (WSIs), yet it often encounters overfitting challenges in real-world applications. This paper reveals the correlation between MIL's performance and the entropy of attention values. Based on this observation, we propose Attention Diversity Regularization (ADR), a simple but effective technique aimed at promoting high entropy in attention values. Specifically, ADR introduces a negative Shannon entropy loss for attention values into the regular MIL framework. Compared to existing methods aimed at alleviating overfitting, which often necessitate additional modules or processing steps, our ADR approach requires no such extras, demonstrating simplicity and efficiency. We evaluate our ADR on three WSI classification tasks. ADR achieves superior performance over the state-of-the-art on most of them. We also show that ADR can enhance heatmaps, aligning them better with pathologists' diagnostic criteria. The source code is available at \url{https://github.com/dazhangyu123/ADR}.
Benchmarking PathCLIP for Pathology Image Analysis
Jan 05, 2024



Accurate image classification and retrieval are of importance for clinical diagnosis and treatment decision-making. The recent contrastive language-image pretraining (CLIP) model has shown remarkable proficiency in understanding natural images. Drawing inspiration from CLIP, PathCLIP is specifically designed for pathology image analysis, utilizing over 200,000 image and text pairs in training. While the performance the PathCLIP is impressive, its robustness under a wide range of image corruptions remains unknown. Therefore, we conduct an extensive evaluation to analyze the performance of PathCLIP on various corrupted images from the datasets of Osteosarcoma and WSSS4LUAD. In our experiments, we introduce seven corruption types including brightness, contrast, Gaussian blur, resolution, saturation, hue, and markup at four severity levels. Through experiments, we find that PathCLIP is relatively robustness to image corruptions and surpasses OpenAI-CLIP and PLIP in zero-shot classification. Among the seven corruptions, blur and resolution can cause server performance degradation of the PathCLIP. This indicates that ensuring the quality of images is crucial before conducting a clinical test. Additionally, we assess the robustness of PathCLIP in the task of image-image retrieval, revealing that PathCLIP performs less effectively than PLIP on Osteosarcoma but performs better on WSSS4LUAD under diverse corruptions. Overall, PathCLIP presents impressive zero-shot classification and retrieval performance for pathology images, but appropriate care needs to be taken when using it. We hope this study provides a qualitative impression of PathCLIP and helps understand its differences from other CLIP models.