 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCPathAgent: An Agent-based Foundation Model for Interpretable High-Resolution Pathology Image Analysis Mimicking Pathologists' Diagnostic Logic
May 26, 2025Recent advances in computational pathology have led to the emergence of numerous foundation models. However, these approaches fail to replicate the diagnostic process of pathologists, as they either simply rely on general-purpose encoders with multi-instance learning for classification or directly apply multimodal models to generate reports from images. A significant limitation is their inability to emulate the diagnostic logic employed by pathologists, who systematically examine slides at low magnification for overview before progressively zooming in on suspicious regions to formulate comprehensive diagnoses. To address this gap, we introduce CPathAgent, an innovative agent-based model that mimics pathologists' reasoning processes by autonomously executing zoom-in/out and navigation operations across pathology images based on observed visual features. To achieve this, we develop a multi-stage training strategy unifying patch-level, region-level, and whole-slide capabilities within a single model, which is essential for mimicking pathologists, who require understanding and reasoning capabilities across all three scales. This approach generates substantially more detailed and interpretable diagnostic reports compared to existing methods, particularly for huge region understanding. Additionally, we construct an expert-validated PathMMU-HR$^{2}$, the first benchmark for huge region analysis, a critical intermediate scale between patches and whole slides, as diagnosticians typically examine several key regions rather than entire slides at once. Extensive experiments demonstrate that CPathAgent consistently outperforms existing approaches across three scales of benchmarks, validating the effectiveness of our agent-based diagnostic approach and highlighting a promising direction for the future development of computational pathology.
PathVQ: Reforming Computational Pathology Foundation Model for Whole Slide Image Analysis via Vector Quantization
Mar 09, 2025Computational pathology and whole-slide image (WSI) analysis are pivotal in cancer diagnosis and prognosis. However, the ultra-high resolution of WSIs presents significant modeling challenges. Recent advancements in pathology foundation models have improved performance, yet most approaches rely on [CLS] token representation of tile ViT as slide-level inputs (16x16 pixels is refereed as patch and 224x224 pixels as tile). This discards critical spatial details from patch tokens, limiting downstream WSI analysis tasks. We find that leveraging all spatial patch tokens benefits WSI analysis but incurs nearly 200x higher storage and training costs (e.g., 196 tokens in ViT$_{224}$). To address this, we introduce vector quantized (VQ) distillation on patch feature, which efficiently compresses spatial patch tokens using discrete indices and a decoder. Our method reduces token dimensionality from 1024 to 16, achieving a 64x compression rate while preserving reconstruction fidelity. Furthermore, we employ a multi-scale VQ (MSVQ) strategy, which not only enhances VQ reconstruction performance but also serves as a Self-supervised Learning (SSL) supervision for a seamless slide-level pretraining objective. Built upon the quantized patch features and supervision targets of tile via MSVQ, we develop a progressive convolutional module and slide-level SSL to extract representations with rich spatial-information for downstream WSI tasks. Extensive evaluations on multiple datasets demonstrate the effectiveness of our approach, achieving state-of-the-art performance in WSI analysis. Code will be available soon.
Towards Effective and Efficient Context-aware Nucleus Detection in Histopathology Whole Slide Images
Mar 04, 2025
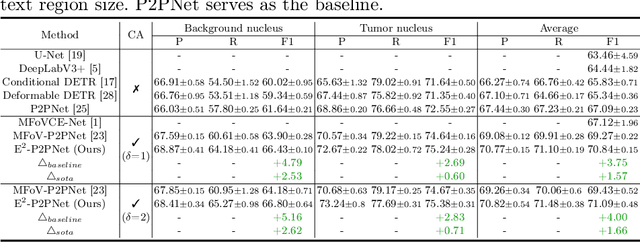
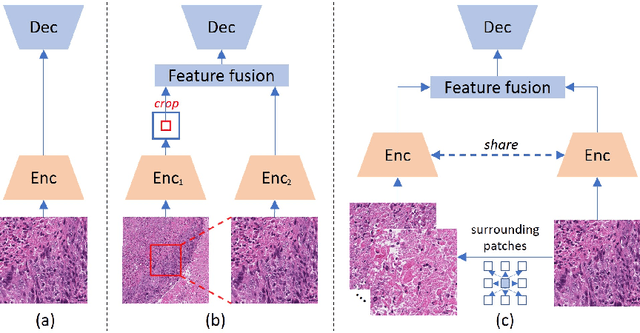
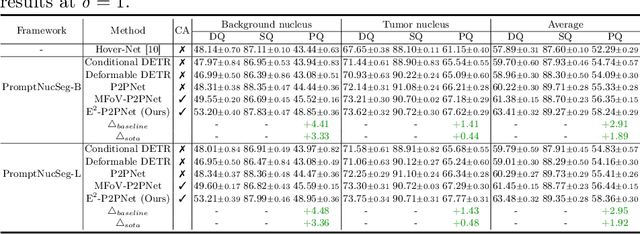
Nucleus detection in histopathology whole slide images (WSIs) is crucial for a broad spectrum of clinical applications. Current approaches for nucleus detection in gigapixel WSIs utilize a sliding window methodology, which overlooks boarder contextual information (eg, tissue structure) and easily leads to inaccurate predictions. To address this problem, recent studies additionally crops a large Filed-of-View (FoV) region around each sliding window to extract contextual features. However, such methods substantially increases the inference latency. In this paper, we propose an effective and efficient context-aware nucleus detection algorithm. Specifically, instead of leveraging large FoV regions, we aggregate contextual clues from off-the-shelf features of historically visited sliding windows. This design greatly reduces computational overhead. Moreover, compared to large FoV regions at a low magnification, the sliding window patches have higher magnification and provide finer-grained tissue details, thereby enhancing the detection accuracy. To further improve the efficiency, we propose a grid pooling technique to compress dense feature maps of each patch into a few contextual tokens. Finally, we craft OCELOT-seg, the first benchmark dedicated to context-aware nucleus instance segmentation. Code, dataset, and model checkpoints will be available at https://github.com/windygoo/PathContext.
Large-scale and Fine-grained Vision-language Pre-training for Enhanced CT Image Understanding
Jan 24, 2025



Artificial intelligence (AI) shows great potential in assisting radiologists to improve the efficiency and accuracy of medical image interpretation and diagnosis. However, a versatile AI model requires large-scale data and comprehensive annotations, which are often impractical in medical settings. Recent studies leverage radiology reports as a naturally high-quality supervision for medical images, using contrastive language-image pre-training (CLIP) to develop language-informed models for radiological image interpretation. Nonetheless, these approaches typically contrast entire images with reports, neglecting the local associations between imaging regions and report sentences, which may undermine model performance and interoperability. In this paper, we propose a fine-grained vision-language model (fVLM) for anatomy-level CT image interpretation. Specifically, we explicitly match anatomical regions of CT images with corresponding descriptions in radiology reports and perform contrastive pre-training for each anatomy individually. Fine-grained alignment, however, faces considerable false-negative challenges, mainly from the abundance of anatomy-level healthy samples and similarly diseased abnormalities. To tackle this issue, we propose identifying false negatives of both normal and abnormal samples and calibrating contrastive learning from patient-level to disease-aware pairing. We curated the largest CT dataset to date, comprising imaging and report data from 69,086 patients, and conducted a comprehensive evaluation of 54 major and important disease diagnosis tasks across 15 main anatomies. Experimental results demonstrate the substantial potential of fVLM in versatile medical image interpretation. In the zero-shot classification task, we achieved an average AUC of 81.3% on 54 diagnosis tasks, surpassing CLIP and supervised methods by 12.9% and 8.0%, respectively.
CPath-Omni: A Unified Multimodal Foundation Model for Patch and Whole Slide Image Analysis in Computational Pathology
Dec 16, 2024The emergence of large multimodal models (LMMs) has brought significant advancements to pathology. Previous research has primarily focused on separately training patch-level and whole-slide image (WSI)-level models, limiting the integration of learned knowledge across patches and WSIs, and resulting in redundant models. In this work, we introduce CPath-Omni, the first 15-billion-parameter LMM designed to unify both patch and WSI level image analysis, consolidating a variety of tasks at both levels, including classification, visual question answering, captioning, and visual referring prompting. Extensive experiments demonstrate that CPath-Omni achieves state-of-the-art (SOTA) performance across seven diverse tasks on 39 out of 42 datasets, outperforming or matching task-specific models trained for individual tasks. Additionally, we develop a specialized pathology CLIP-based visual processor for CPath-Omni, CPath-CLIP, which, for the first time, integrates different vision models and incorporates a large language model as a text encoder to build a more powerful CLIP model, which achieves SOTA performance on nine zero-shot and four few-shot datasets. Our findings highlight CPath-Omni's ability to unify diverse pathology tasks, demonstrating its potential to streamline and advance the field of foundation model in pathology.
Rethinking Transformer for Long Contextual Histopathology Whole Slide Image Analysis
Oct 18, 2024



Histopathology Whole Slide Image (WSI) analysis serves as the gold standard for clinical cancer diagnosis in the daily routines of doctors. To develop computer-aided diagnosis model for WSIs, previous methods typically employ Multi-Instance Learning to enable slide-level prediction given only slide-level labels. Among these models, vanilla attention mechanisms without pairwise interactions have traditionally been employed but are unable to model contextual information. More recently, self-attention models have been utilized to address this issue. To alleviate the computational complexity of long sequences in large WSIs, methods like HIPT use region-slicing, and TransMIL employs approximation of full self-attention. Both approaches suffer from suboptimal performance due to the loss of key information. Moreover, their use of absolute positional embedding struggles to effectively handle long contextual dependencies in shape-varying WSIs. In this paper, we first analyze how the low-rank nature of the long-sequence attention matrix constrains the representation ability of WSI modelling. Then, we demonstrate that the rank of attention matrix can be improved by focusing on local interactions via a local attention mask. Our analysis shows that the local mask aligns with the attention patterns in the lower layers of the Transformer. Furthermore, the local attention mask can be implemented during chunked attention calculation, reducing the quadratic computational complexity to linear with a small local bandwidth. Building on this, we propose a local-global hybrid Transformer for both computational acceleration and local-global information interactions modelling. Our method, Long-contextual MIL (LongMIL), is evaluated through extensive experiments on various WSI tasks to validate its superiority. Our code will be available at github.com/invoker-LL/Long-MIL.
Large-scale cervical precancerous screening via AI-assisted cytology whole slide image analysis
Jul 28, 2024



Cervical Cancer continues to be the leading gynecological malignancy, posing a persistent threat to women's health on a global scale. Early screening via cytology Whole Slide Image (WSI) diagnosis is critical to prevent this Cancer progression and improve survival rate, but pathologist's single test suffers inevitable false negative due to the immense number of cells that need to be reviewed within a WSI. Though computer-aided automated diagnostic models can serve as strong complement for pathologists, their effectiveness is hampered by the paucity of extensive and detailed annotations, coupled with the limited interpretability and robustness. These factors significantly hinder their practical applicability and reliability in clinical settings. To tackle these challenges, we develop an AI approach, which is a Scalable Technology for Robust and Interpretable Diagnosis built on Extensive data (STRIDE) of cervical cytology. STRIDE addresses the bottleneck of limited annotations by integrating patient-level labels with a small portion of cell-level labels through an end-to-end training strategy, facilitating scalable learning across extensive datasets. To further improve the robustness to real-world domain shifts of cytology slide-making and imaging, STRIDE employs color adversarial samples training that mimic staining and imaging variations. Lastly, to achieve pathologist-level interpretability for the trustworthiness in clinical settings, STRIDE can generate explanatory textual descriptions that simulates pathologists' diagnostic processes by cell image feature and textual description alignment. Conducting extensive experiments and evaluations in 183 medical centers with a dataset of 341,889 WSIs and 0.1 billion cells from cervical cytology patients, STRIDE has demonstrated a remarkable superiority over previous state-of-the-art techniques.
PathGen-1.6M: 1.6 Million Pathology Image-text Pairs Generation through Multi-agent Collaboration
Jun 28, 2024



Vision Language Models (VLMs) like CLIP have attracted substantial attention in pathology, serving as backbones for applications such as zero-shot image classification and Whole Slide Image (WSI) analysis. Additionally, they can function as vision encoders when combined with large language models (LLMs) to support broader capabilities. Current efforts to train pathology VLMs rely on pathology image-text pairs from platforms like PubMed, YouTube, and Twitter, which provide limited, unscalable data with generally suboptimal image quality. In this work, we leverage large-scale WSI datasets like TCGA to extract numerous high-quality image patches. We then train a large multimodal model to generate captions for these images, creating PathGen-1.6M, a dataset containing 1.6 million high-quality image-caption pairs. Our approach involves multiple agent models collaborating to extract representative WSI patches, generating and refining captions to obtain high-quality image-text pairs. Extensive experiments show that integrating these generated pairs with existing datasets to train a pathology-specific CLIP model, PathGen-CLIP, significantly enhances its ability to analyze pathological images, with substantial improvements across nine pathology-related zero-shot image classification tasks and three whole-slide image tasks. Furthermore, we construct 200K instruction-tuning data based on PathGen-1.6M and integrate PathGen-CLIP with the Vicuna LLM to create more powerful multimodal models through instruction tuning. Overall, we provide a scalable pathway for high-quality data generation in pathology, paving the way for next-generation general pathology models.
ADR: Attention Diversification Regularization for Mitigating Overfitting in Multiple Instance Learning based Whole Slide Image Classification
Jun 18, 2024



Multiple Instance Learning (MIL) has demonstrated effectiveness in analyzing whole slide images (WSIs), yet it often encounters overfitting challenges in real-world applications. This paper reveals the correlation between MIL's performance and the entropy of attention values. Based on this observation, we propose Attention Diversity Regularization (ADR), a simple but effective technique aimed at promoting high entropy in attention values. Specifically, ADR introduces a negative Shannon entropy loss for attention values into the regular MIL framework. Compared to existing methods aimed at alleviating overfitting, which often necessitate additional modules or processing steps, our ADR approach requires no such extras, demonstrating simplicity and efficiency. We evaluate our ADR on three WSI classification tasks. ADR achieves superior performance over the state-of-the-art on most of them. We also show that ADR can enhance heatmaps, aligning them better with pathologists' diagnostic criteria. The source code is available at \url{https://github.com/dazhangyu123/ADR}.
Multi-modal Learning with Missing Modality in Predicting Axillary Lymph Node Metastasis
Jan 03, 2024



Multi-modal Learning has attracted widespread attention in medical image analysis. Using multi-modal data, whole slide images (WSIs) and clinical information, can improve the performance of deep learning models in the diagnosis of axillary lymph node metastasis. However, clinical information is not easy to collect in clinical practice due to privacy concerns, limited resources, lack of interoperability, etc. Although patient selection can ensure the training set to have multi-modal data for model development, missing modality of clinical information can appear during test. This normally leads to performance degradation, which limits the use of multi-modal models in the clinic. To alleviate this problem, we propose a bidirectional distillation framework consisting of a multi-modal branch and a single-modal branch. The single-modal branch acquires the complete multi-modal knowledge from the multi-modal branch, while the multi-modal learns the robust features of WSI from the single-modal. We conduct experiments on a public dataset of Lymph Node Metastasis in Early Breast Cancer to validate the method. Our approach not only achieves state-of-the-art performance with an AUC of 0.861 on the test set without missing data, but also yields an AUC of 0.842 when the rate of missing modality is 80\%. This shows the effectiveness of the approach in dealing with multi-modal data and missing modality. Such a model has the potential to improve treatment decision-making for early breast cancer patients who have axillary lymph node metastatic status.