 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHierarchical Graph Capsule Network
Dec 16, 2020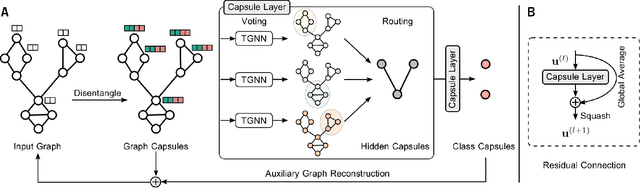
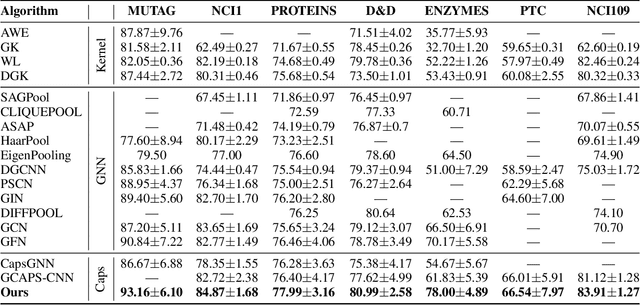
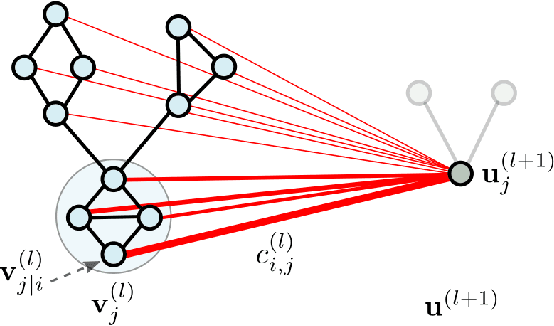
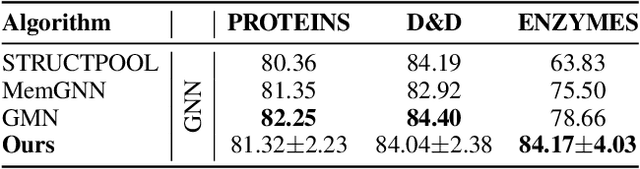
Graph Neural Networks (GNNs) draw their strength from explicitly modeling the topological information of structured data. However, existing GNNs suffer from limited capability in capturing the hierarchical graph representation which plays an important role in graph classification. In this paper, we innovatively propose hierarchical graph capsule network (HGCN) that can jointly learn node embeddings and extract graph hierarchies. Specifically, disentangled graph capsules are established by identifying heterogeneous factors underlying each node, such that their instantiation parameters represent different properties of the same entity. To learn the hierarchical representation, HGCN characterizes the part-whole relationship between lower-level capsules (part) and higher-level capsules (whole) by explicitly considering the structure information among the parts. Experimental studies demonstrate the effectiveness of HGCN and the contribution of each component.
Deep Multimodal Fusion by Channel Exchanging
Nov 10, 2020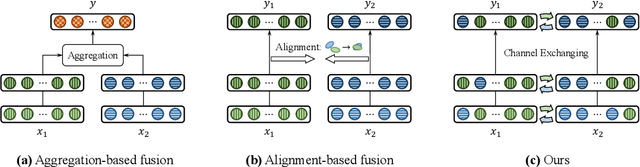
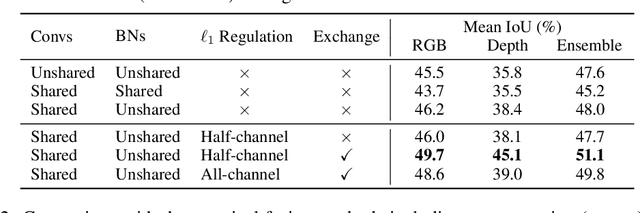
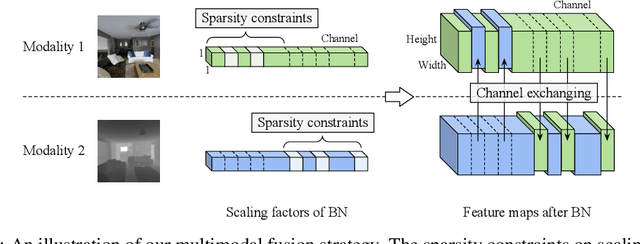
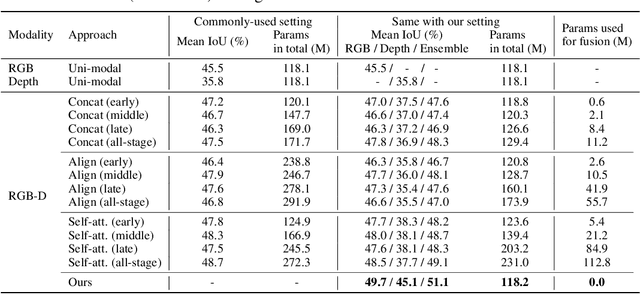
Deep multimodal fusion by using multiple sources of data for classification or regression has exhibited a clear advantage over the unimodal counterpart on various applications. Yet, current methods including aggregation-based and alignment-based fusion are still inadequate in balancing the trade-off between inter-modal fusion and intra-modal processing, incurring a bottleneck of performance improvement. To this end, this paper proposes Channel-Exchanging-Network (CEN), a parameter-free multimodal fusion framework that dynamically exchanges channels between sub-networks of different modalities. Specifically, the channel exchanging process is self-guided by individual channel importance that is measured by the magnitude of Batch-Normalization (BN) scaling factor during training. The validity of such exchanging process is also guaranteed by sharing convolutional filters yet keeping separate BN layers across modalities, which, as an add-on benefit, allows our multimodal architecture to be almost as compact as a unimodal network. Extensive experiments on semantic segmentation via RGB-D data and image translation through multi-domain input verify the effectiveness of our CEN compared to current state-of-the-art methods. Detailed ablation studies have also been carried out, which provably affirm the advantage of each component we propose. Our code is available at https://github.com/yikaiw/CEN.
On Self-Distilling Graph Neural Network
Nov 04, 2020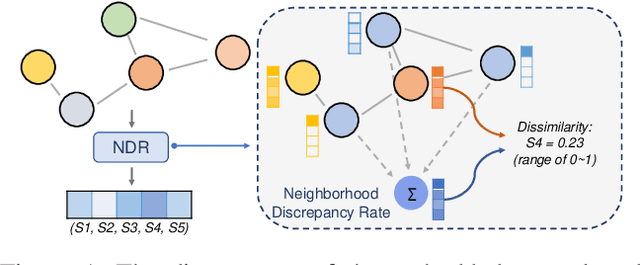
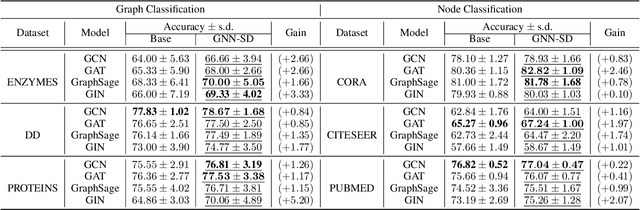
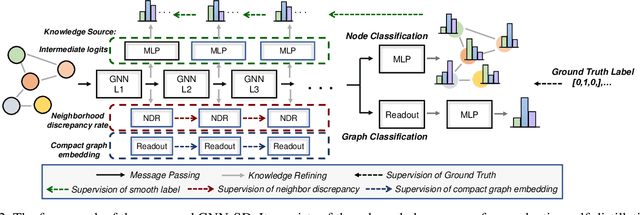
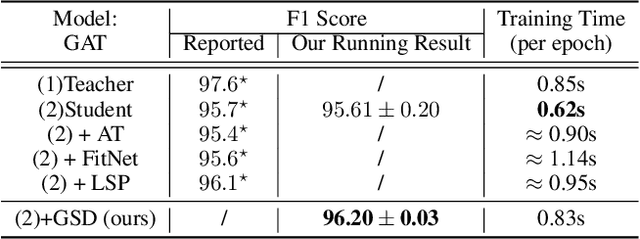
Recently, the teacher-student knowledge distillation framework has demonstrated its potential in training Graph Neural Networks (GNNs). However, due to the difficulty of training deep and wide GNN models, one can not always obtain a satisfactory teacher model for distillation. Furthermore, the inefficient training process of teacher-student knowledge distillation also impedes its applications in GNN models. In this paper, we propose the first teacher-free knowledge distillation framework for GNNs, termed GNN Self-Distillation (GNN-SD), that serves as a drop-in replacement for improving the training process of GNNs.We design three knowledge sources for GNN-SD: neighborhood discrepancy rate (NDR), compact graph embedding and intermediate logits. Notably, serving as a metric of the non-smoothness of the embedded graph, NDR empowers the transferability of knowledge that maintains high neighborhood discrepancy by enforcing consistency between consecutive GNN layers. We conduct exploring analysis to verify that our framework could improve the training dynamics and embedding quality of GNNs. Extensive experiments on various popular GNN models and datasets demonstrate that our approach obtains consistent and considerable performance enhancement, proving its effectiveness and generalization ability.
RetroXpert: Decompose Retrosynthesis Prediction like a Chemist
Nov 04, 2020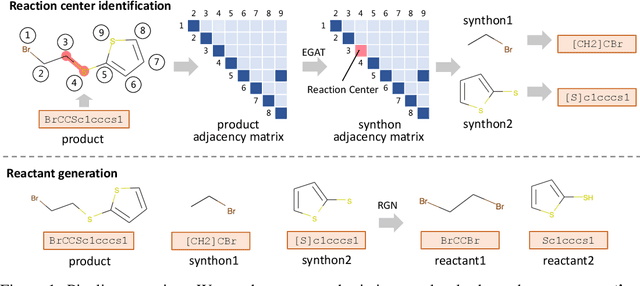
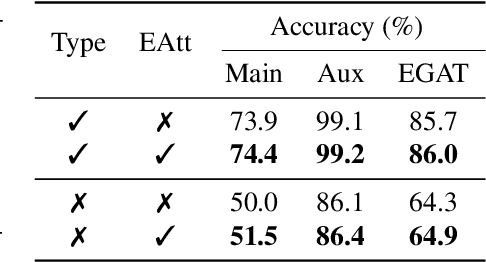
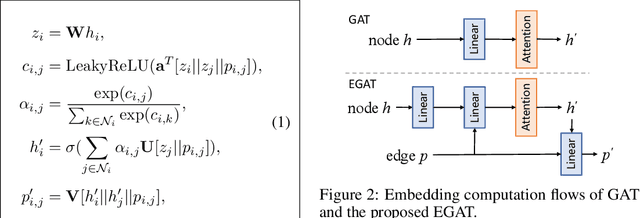
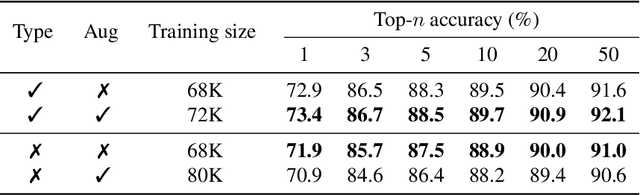
Retrosynthesis is the process of recursively decomposing target molecules into available building blocks. It plays an important role in solving problems in organic synthesis planning. To automate or assist in the retrosynthesis analysis, various retrosynthesis prediction algorithms have been proposed. However, most of them are cumbersome and lack interpretability about their predictions. In this paper, we devise a novel template-free algorithm for automatic retrosynthetic expansion inspired by how chemists approach retrosynthesis prediction. Our method disassembles retrosynthesis into two steps: i) identify the potential reaction center of the target molecule through a novel graph neural network and generate intermediate synthons, and ii) generate the reactants associated with synthons via a robust reactant generation model. While outperforming the state-of-the-art baselines by a significant margin, our model also provides chemically reasonable interpretation.
Graph Information Bottleneck for Subgraph Recognition
Oct 12, 2020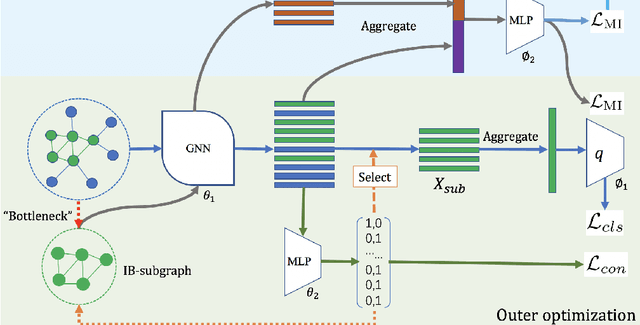
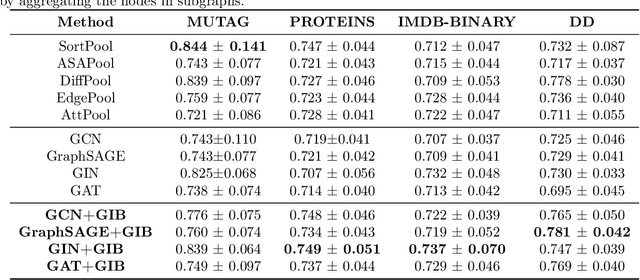
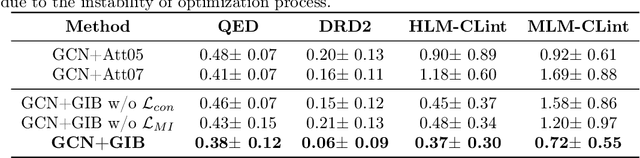
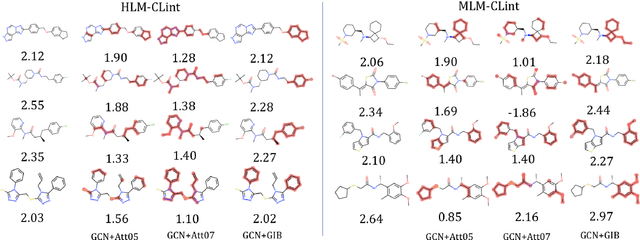
Given the input graph and its label/property, several key problems of graph learning, such as finding interpretable subgraphs, graph denoising and graph compression, can be attributed to the fundamental problem of recognizing a subgraph of the original one. This subgraph shall be as informative as possible, yet contains less redundant and noisy structure. This problem setting is closely related to the well-known information bottleneck (IB) principle, which, however, has less been studied for the irregular graph data and graph neural networks (GNNs). In this paper, we propose a framework of Graph Information Bottleneck (GIB) for the subgraph recognition problem in deep graph learning. Under this framework, one can recognize the maximally informative yet compressive subgraph, named IB-subgraph. However, the GIB objective is notoriously hard to optimize, mostly due to the intractability of the mutual information of irregular graph data and the unstable optimization process. In order to tackle these challenges, we propose: i) a GIB objective based-on a mutual information estimator for the irregular graph data; ii) a bi-level optimization scheme to maximize the GIB objective; iii) a connectivity loss to stabilize the optimization process. We evaluate the properties of the IB-subgraph in three application scenarios: improvement of graph classification, graph interpretation and graph denoising. Extensive experiments demonstrate that the information-theoretic IB-subgraph enjoys superior graph properties.
Whole Slide Images based Cancer Survival Prediction using Attention Guided Deep Multiple Instance Learning Networks
Sep 23, 2020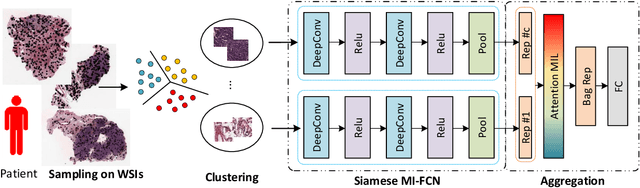
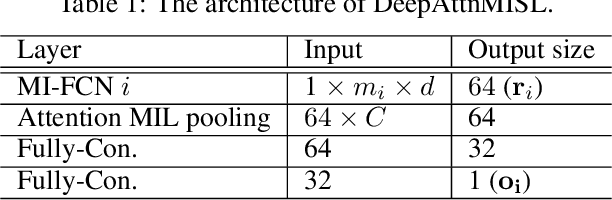

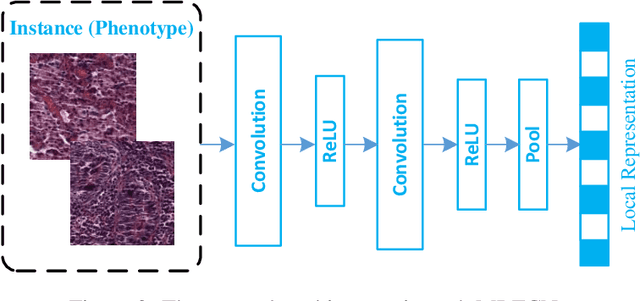
Traditional image-based survival prediction models rely on discriminative patch labeling which make those methods not scalable to extend to large datasets. Recent studies have shown Multiple Instance Learning (MIL) framework is useful for histopathological images when no annotations are available in classification task. Different to the current image-based survival models that limit to key patches or clusters derived from Whole Slide Images (WSIs), we propose Deep Attention Multiple Instance Survival Learning (DeepAttnMISL) by introducing both siamese MI-FCN and attention-based MIL pooling to efficiently learn imaging features from the WSI and then aggregate WSI-level information to patient-level. Attention-based aggregation is more flexible and adaptive than aggregation techniques in recent survival models. We evaluated our methods on two large cancer whole slide images datasets and our results suggest that the proposed approach is more effective and suitable for large datasets and has better interpretability in locating important patterns and features that contribute to accurate cancer survival predictions. The proposed framework can also be used to assess individual patient's risk and thus assisting in delivering personalized medicine. Codes are available at https://github.com/uta-smile/DeepAttnMISL_MEDIA.
* 22 pages, 13 figures, published in Medical Image Analysis 65, 101789
Microscope Based HER2 Scoring System
Sep 15, 2020

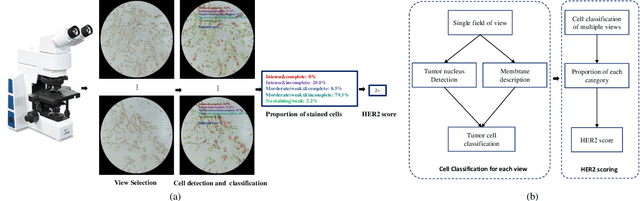
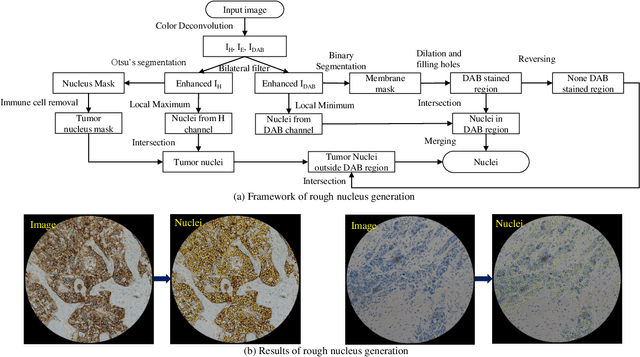
The overexpression of human epidermal growth factor receptor 2 (HER2) has been established as a therapeutic target in multiple types of cancers, such as breast and gastric cancers. Immunohistochemistry (IHC) is employed as a basic HER2 test to identify the HER2-positive, borderline, and HER2-negative patients. However, the reliability and accuracy of HER2 scoring are affected by many factors, such as pathologists' experience. Recently, artificial intelligence (AI) has been used in various disease diagnosis to improve diagnostic accuracy and reliability, but the interpretation of diagnosis results is still an open problem. In this paper, we propose a real-time HER2 scoring system, which follows the HER2 scoring guidelines to complete the diagnosis, and thus each step is explainable. Unlike the previous scoring systems based on whole-slide imaging, our HER2 scoring system is integrated into an augmented reality (AR) microscope that can feedback AI results to the pathologists while reading the slide. The pathologists can help select informative fields of view (FOVs), avoiding the confounding regions, such as DCIS. Importantly, we illustrate the intermediate results with membrane staining condition and cell classification results, making it possible to evaluate the reliability of the diagnostic results. Also, we support the interactive modification of selecting regions-of-interest, making our system more flexible in clinical practice. The collaboration of AI and pathologists can significantly improve the robustness of our system. We evaluate our system with 285 breast IHC HER2 slides, and the classification accuracy of 95\% shows the effectiveness of our HER2 scoring system.
Tackling Over-Smoothing for General Graph Convolutional Networks
Sep 08, 2020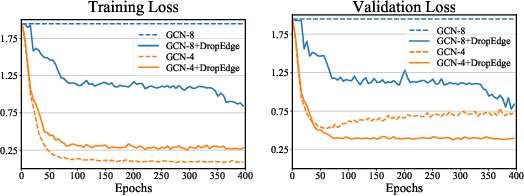
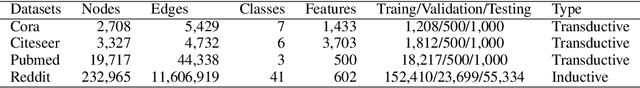
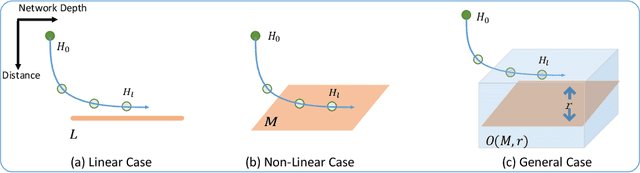
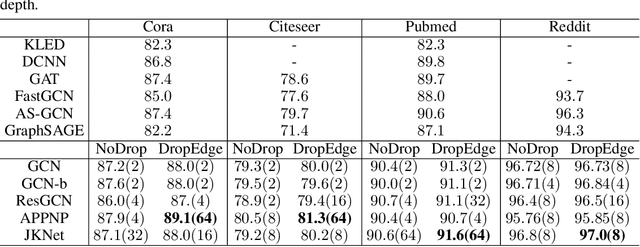
Increasing the depth of GCN, which is expected to permit more expressivity, is shown to incur performance detriment especially on node classification. The main cause of this lies in over-smoothing. The over-smoothing issue drives the output of GCN towards a space that contains limited distinguished information among nodes, leading to poor expressivity. Several works on refining the architecture of deep GCN have been proposed, but it is still unknown in theory whether or not these refinements are able to relieve over-smoothing. In this paper, we first theoretically analyze how general GCNs act with the increase in depth, including generic GCN, GCN with bias, ResGCN, and APPNP. We find that all these models are characterized by a universal process: all nodes converging to a cuboid. Upon this theorem, we propose DropEdge to alleviate over-smoothing by randomly removing a certain number of edges at each training epoch. Theoretically, DropEdge either reduces the convergence speed of over-smoothing or relieves the information loss caused by dimension collapse. Experimental evaluations on simulated dataset have visualized the difference in over-smoothing between different GCNs. Moreover, extensive experiments on several real benchmarks support that DropEdge consistently improves the performance on a variety of both shallow and deep GCNs.
User-Guided Domain Adaptation for Rapid Annotation from User Interactions: A Study on Pathological Liver Segmentation
Sep 05, 2020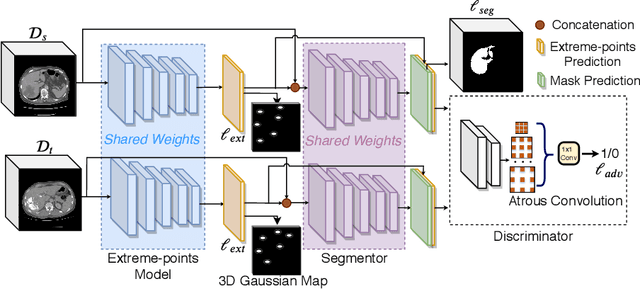
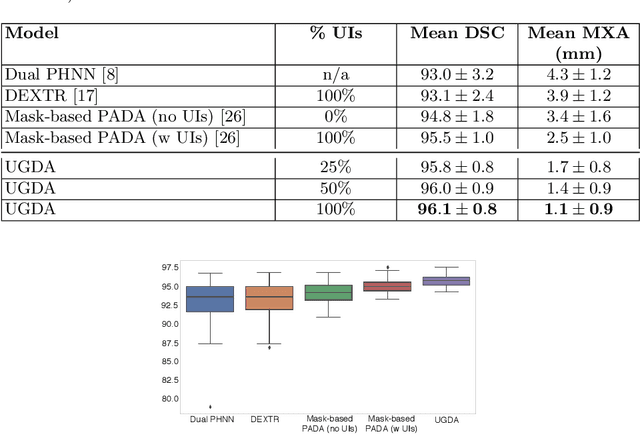


Mask-based annotation of medical images, especially for 3D data, is a bottleneck in developing reliable machine learning models. Using minimal-labor user interactions (UIs) to guide the annotation is promising, but challenges remain on best harmonizing the mask prediction with the UIs. To address this, we propose the user-guided domain adaptation (UGDA) framework, which uses prediction-based adversarial domain adaptation (PADA) to model the combined distribution of UIs and mask predictions. The UIs are then used as anchors to guide and align the mask prediction. Importantly, UGDA can both learn from unlabelled data and also model the high-level semantic meaning behind different UIs. We test UGDA on annotating pathological livers using a clinically comprehensive dataset of 927 patient studies. Using only extreme-point UIs, we achieve a mean (worst-case) performance of 96.1%(94.9%), compared to 93.0% (87.0%) for deep extreme points (DEXTR). Furthermore, we also show UGDA can retain this state-of-the-art performance even when only seeing a fraction of available UIs, demonstrating an ability for robust and reliable UI-guided segmentation with extremely minimal labor demands.
Improving Generative Adversarial Networks with Local Coordinate Coding
Jul 28, 2020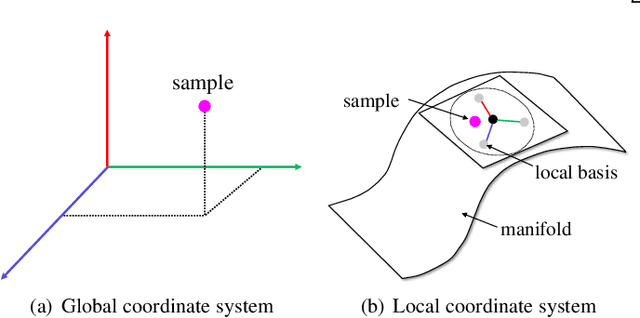
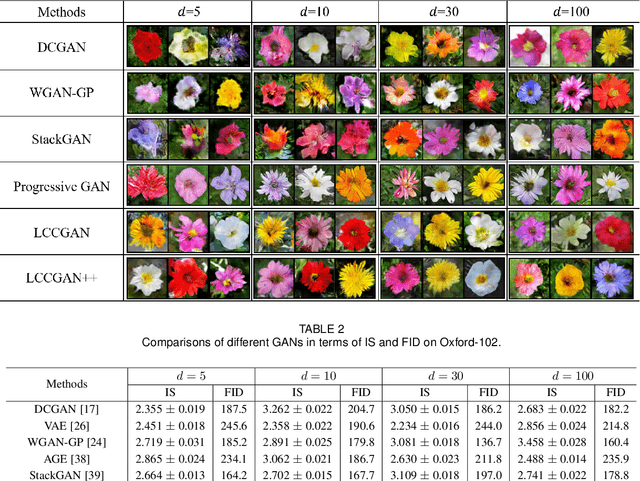
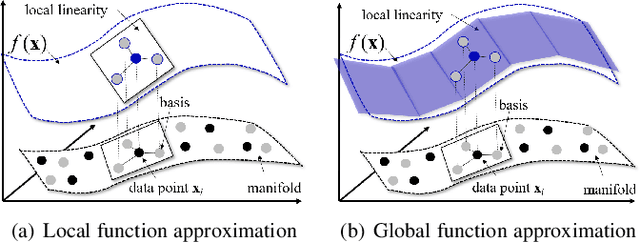
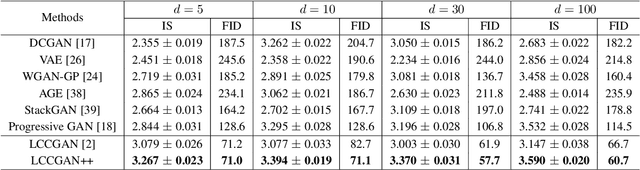
Generative adversarial networks (GANs) have shown remarkable success in generating realistic data from some predefined prior distribution (e.g., Gaussian noises). However, such prior distribution is often independent of real data and thus may lose semantic information (e.g., geometric structure or content in images) of data. In practice, the semantic information might be represented by some latent distribution learned from data. However, such latent distribution may incur difficulties in data sampling for GANs. In this paper, rather than sampling from the predefined prior distribution, we propose an LCCGAN model with local coordinate coding (LCC) to improve the performance of generating data. First, we propose an LCC sampling method in LCCGAN to sample meaningful points from the latent manifold. With the LCC sampling method, we can exploit the local information on the latent manifold and thus produce new data with promising quality. Second, we propose an improved version, namely LCCGAN++, by introducing a higher-order term in the generator approximation. This term is able to achieve better approximation and thus further improve the performance. More critically, we derive the generalization bound for both LCCGAN and LCCGAN++ and prove that a low-dimensional input is sufficient to achieve good generalization performance. Extensive experiments on four benchmark datasets demonstrate the superiority of the proposed method over existing GANs.