 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnsupervised Path Representation Learning with Curriculum Negative Sampling
Jun 17, 2021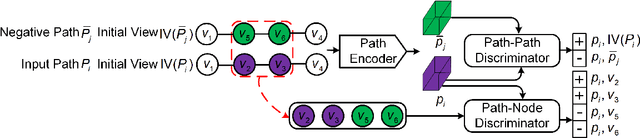
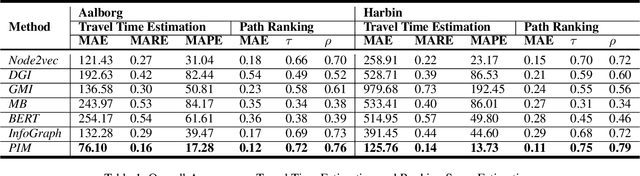
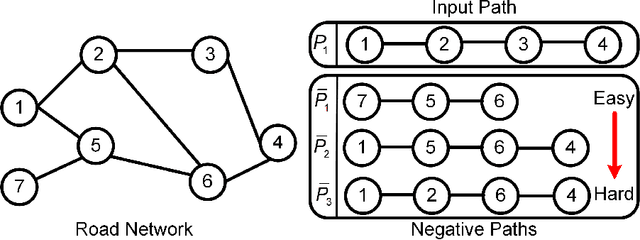
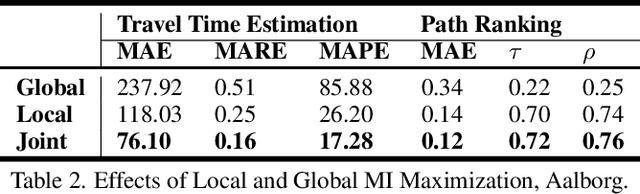
Path representations are critical in a variety of transportation applications, such as estimating path ranking in path recommendation systems and estimating path travel time in navigation systems. Existing studies often learn task-specific path representations in a supervised manner, which require a large amount of labeled training data and generalize poorly to other tasks. We propose an unsupervised learning framework Path InfoMax (PIM) to learn generic path representations that work for different downstream tasks. We first propose a curriculum negative sampling method, for each input path, to generate a small amount of negative paths, by following the principles of curriculum learning. Next, \emph{PIM} employs mutual information maximization to learn path representations from both a global and a local view. In the global view, PIM distinguishes the representations of the input paths from those of the negative paths. In the local view, \emph{PIM} distinguishes the input path representations from the representations of the nodes that appear only in the negative paths. This enables the learned path representations to encode both global and local information at different scales. Extensive experiments on two downstream tasks, ranking score estimation and travel time estimation, using two road network datasets suggest that PIM significantly outperforms other unsupervised methods and is also able to be used as a pre-training method to enhance supervised path representation learning.
Neural Bellman-Ford Networks: A General Graph Neural Network Framework for Link Prediction
Jun 16, 2021
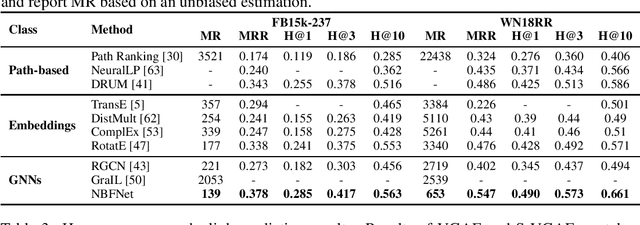
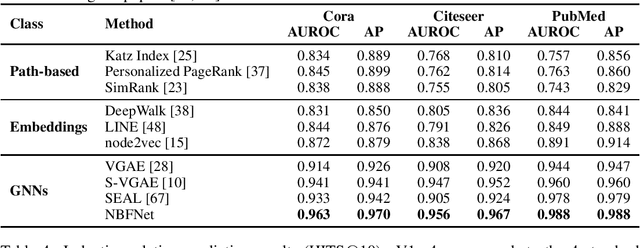
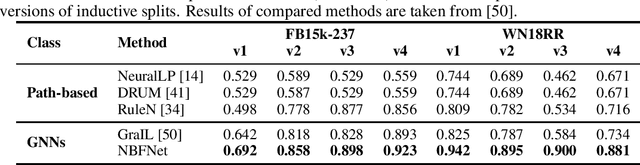
Link prediction is a very fundamental task on graphs. Inspired by traditional path-based methods, in this paper we propose a general and flexible representation learning framework based on paths for link prediction. Specifically, we define the representation of a pair of nodes as the generalized sum of all path representations, with each path representation as the generalized product of the edge representations in the path. Motivated by the Bellman-Ford algorithm for solving the shortest path problem, we show that the proposed path formulation can be efficiently solved by the generalized Bellman-Ford algorithm. To further improve the capacity of the path formulation, we propose the Neural Bellman-Ford Network (NBFNet), a general graph neural network framework that solves the path formulation with learned operators in the generalized Bellman-Ford algorithm. The NBFNet parameterizes the generalized Bellman-Ford algorithm with 3 neural components, namely INDICATOR, MESSAGE and AGGREGATE functions, which corresponds to the boundary condition, multiplication operator, and summation operator respectively. The NBFNet is very general, covers many traditional path-based methods, and can be applied to both homogeneous graphs and multi-relational graphs (e.g., knowledge graphs) in both transductive and inductive settings. Experiments on both homogeneous graphs and knowledge graphs show that the proposed NBFNet outperforms existing methods by a large margin in both transductive and inductive settings, achieving new state-of-the-art results.
Non-Autoregressive Electron Redistribution Modeling for Reaction Prediction
Jun 08, 2021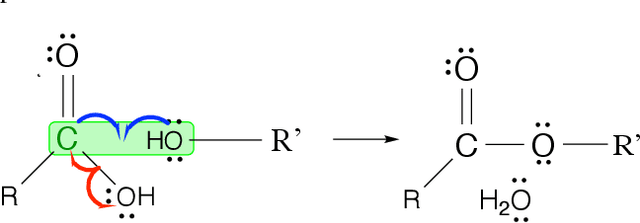
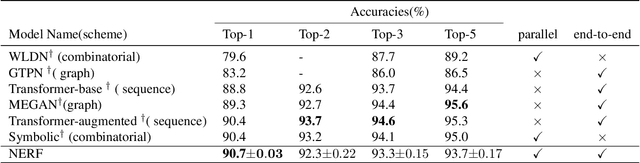
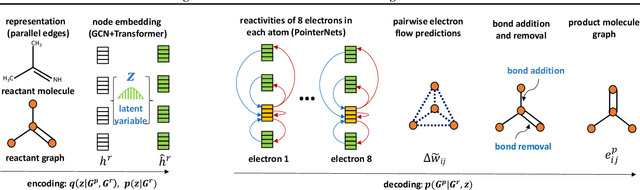
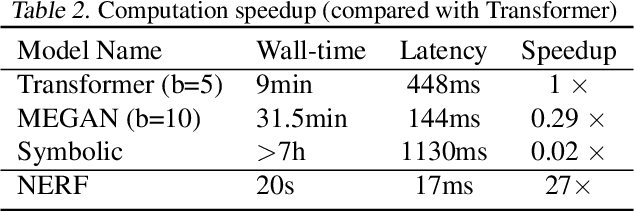
Reliably predicting the products of chemical reactions presents a fundamental challenge in synthetic chemistry. Existing machine learning approaches typically produce a reaction product by sequentially forming its subparts or intermediate molecules. Such autoregressive methods, however, not only require a pre-defined order for the incremental construction but preclude the use of parallel decoding for efficient computation. To address these issues, we devise a non-autoregressive learning paradigm that predicts reaction in one shot. Leveraging the fact that chemical reactions can be described as a redistribution of electrons in molecules, we formulate a reaction as an arbitrary electron flow and predict it with a novel multi-pointer decoding network. Experiments on the USPTO-MIT dataset show that our approach has established a new state-of-the-art top-1 accuracy and achieves at least 27 times inference speedup over the state-of-the-art methods. Also, our predictions are easier for chemists to interpret owing to predicting the electron flows.
Self-supervised Graph-level Representation Learning with Local and Global Structure
Jun 08, 2021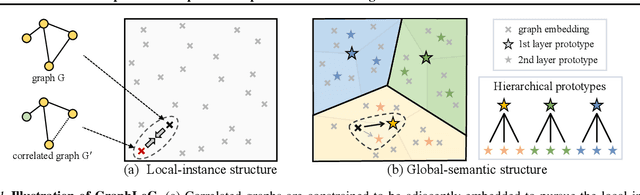
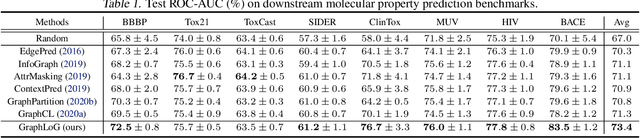
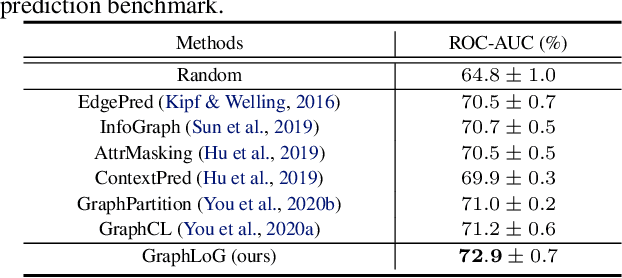

This paper studies unsupervised/self-supervised whole-graph representation learning, which is critical in many tasks such as molecule properties prediction in drug and material discovery. Existing methods mainly focus on preserving the local similarity structure between different graph instances but fail to discover the global semantic structure of the entire data set. In this paper, we propose a unified framework called Local-instance and Global-semantic Learning (GraphLoG) for self-supervised whole-graph representation learning. Specifically, besides preserving the local similarities, GraphLoG introduces the hierarchical prototypes to capture the global semantic clusters. An efficient online expectation-maximization (EM) algorithm is further developed for learning the model. We evaluate GraphLoG by pre-training it on massive unlabeled graphs followed by fine-tuning on downstream tasks. Extensive experiments on both chemical and biological benchmark data sets demonstrate the effectiveness of the proposed approach.
Learning Gradient Fields for Molecular Conformation Generation
Jun 07, 2021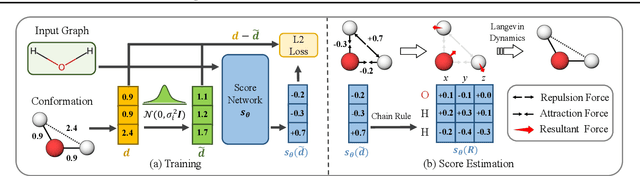
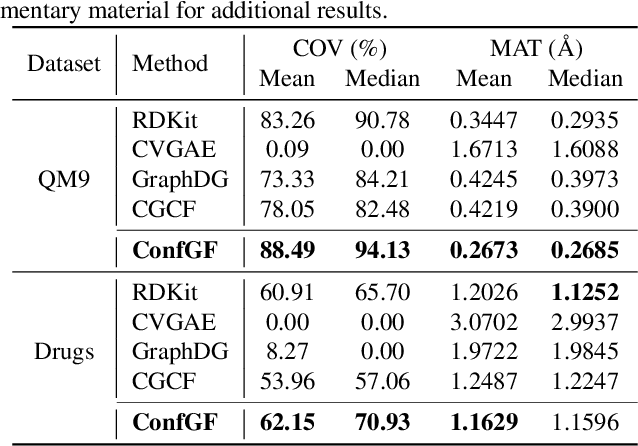
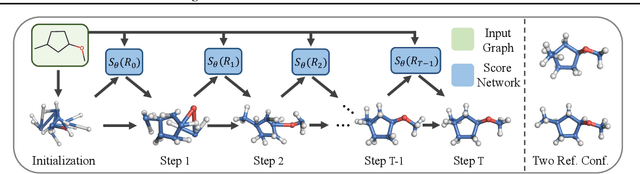
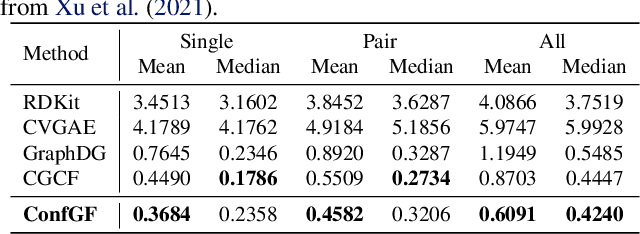
We study a fundamental problem in computational chemistry known as molecular conformation generation, trying to predict stable 3D structures from 2D molecular graphs. Existing machine learning approaches usually first predict distances between atoms and then generate a 3D structure satisfying the distances, where noise in predicted distances may induce extra errors during 3D coordinate generation. Inspired by the traditional force field methods for molecular dynamics simulation, in this paper, we propose a novel approach called ConfGF by directly estimating the gradient fields of the log density of atomic coordinates. The estimated gradient fields allow directly generating stable conformations via Langevin dynamics. However, the problem is very challenging as the gradient fields are roto-translation equivariant. We notice that estimating the gradient fields of atomic coordinates can be translated to estimating the gradient fields of interatomic distances, and hence develop a novel algorithm based on recent score-based generative models to effectively estimate these gradients. Experimental results across multiple tasks show that ConfGF outperforms previous state-of-the-art baselines by a significant margin.
An End-to-End Framework for Molecular Conformation Generation via Bilevel Programming
Jun 02, 2021
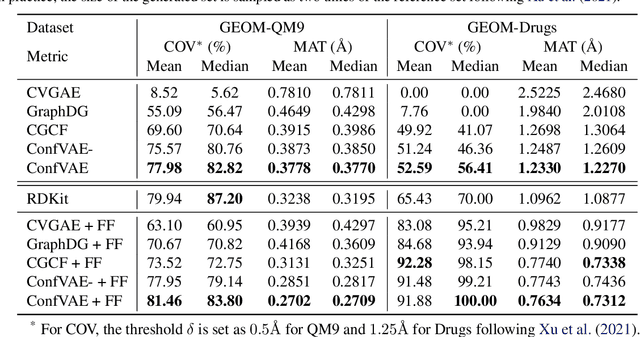
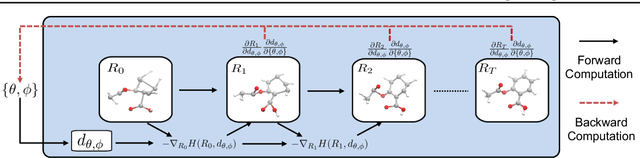
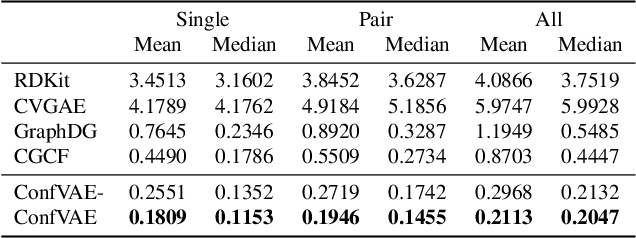
Predicting molecular conformations (or 3D structures) from molecular graphs is a fundamental problem in many applications. Most existing approaches are usually divided into two steps by first predicting the distances between atoms and then generating a 3D structure through optimizing a distance geometry problem. However, the distances predicted with such two-stage approaches may not be able to consistently preserve the geometry of local atomic neighborhoods, making the generated structures unsatisfying. In this paper, we propose an end-to-end solution for molecular conformation prediction called ConfVAE based on the conditional variational autoencoder framework. Specifically, the molecular graph is first encoded in a latent space, and then the 3D structures are generated by solving a principled bilevel optimization program. Extensive experiments on several benchmark data sets prove the effectiveness of our proposed approach over existing state-of-the-art approaches. Code is available at \url{https://github.com/MinkaiXu/ConfVAE-ICML21}.
Learning Neural Generative Dynamics for Molecular Conformation Generation
Feb 28, 2021
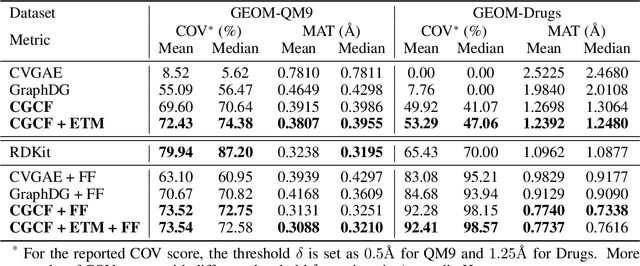
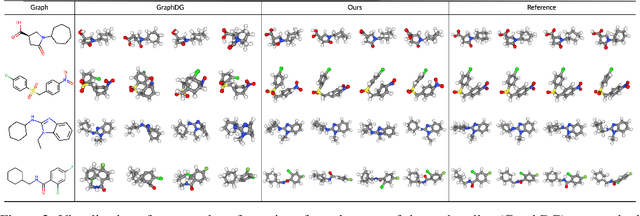
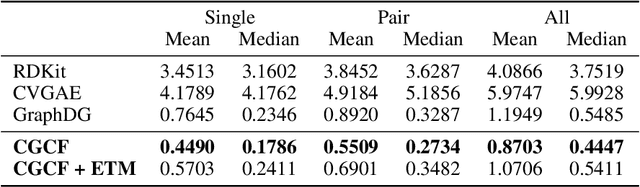
We study how to generate molecule conformations (\textit{i.e.}, 3D structures) from a molecular graph. Traditional methods, such as molecular dynamics, sample conformations via computationally expensive simulations. Recently, machine learning methods have shown great potential by training on a large collection of conformation data. Challenges arise from the limited model capacity for capturing complex distributions of conformations and the difficulty in modeling long-range dependencies between atoms. Inspired by the recent progress in deep generative models, in this paper, we propose a novel probabilistic framework to generate valid and diverse conformations given a molecular graph. We propose a method combining the advantages of both flow-based and energy-based models, enjoying: (1) a high model capacity to estimate the multimodal conformation distribution; (2) explicitly capturing the complex long-range dependencies between atoms in the observation space. Extensive experiments demonstrate the superior performance of the proposed method on several benchmarks, including conformation generation and distance modeling tasks, with a significant improvement over existing generative models for molecular conformation sampling.
Lottery Ticket Implies Accuracy Degradation, Is It a Desirable Phenomenon?
Feb 19, 2021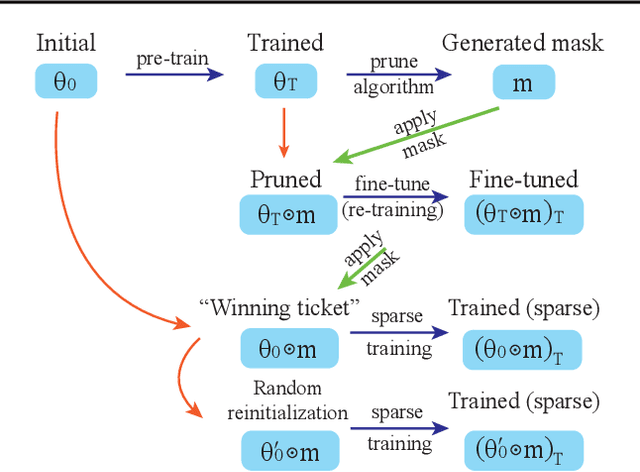
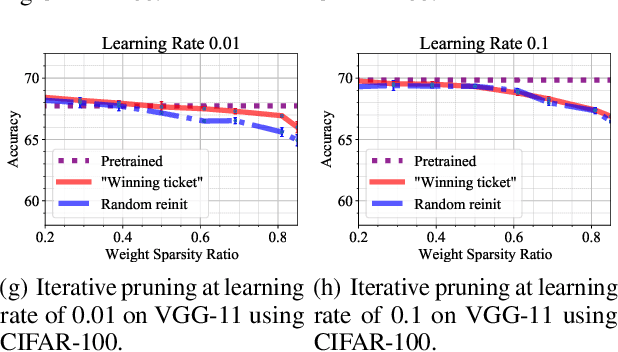
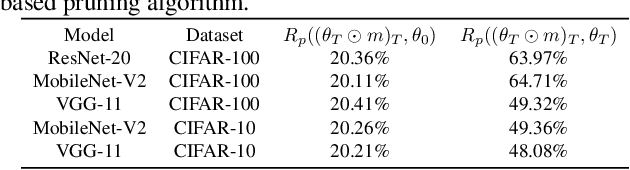
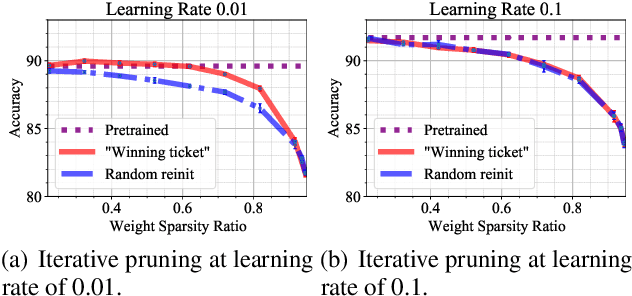
In deep model compression, the recent finding "Lottery Ticket Hypothesis" (LTH) (Frankle & Carbin, 2018) pointed out that there could exist a winning ticket (i.e., a properly pruned sub-network together with original weight initialization) that can achieve competitive performance than the original dense network. However, it is not easy to observe such winning property in many scenarios, where for example, a relatively large learning rate is used even if it benefits training the original dense model. In this work, we investigate the underlying condition and rationale behind the winning property, and find that the underlying reason is largely attributed to the correlation between initialized weights and final-trained weights when the learning rate is not sufficiently large. Thus, the existence of winning property is correlated with an insufficient DNN pretraining, and is unlikely to occur for a well-trained DNN. To overcome this limitation, we propose the "pruning & fine-tuning" method that consistently outperforms lottery ticket sparse training under the same pruning algorithm and the same total training epochs. Extensive experiments over multiple deep models (VGG, ResNet, MobileNet-v2) on different datasets have been conducted to justify our proposals.
Towards Generalized Implementation of Wasserstein Distance in GANs
Jan 12, 2021
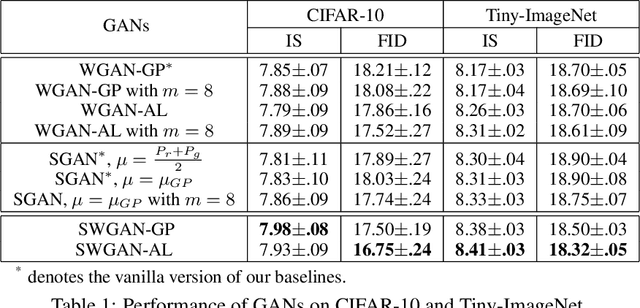
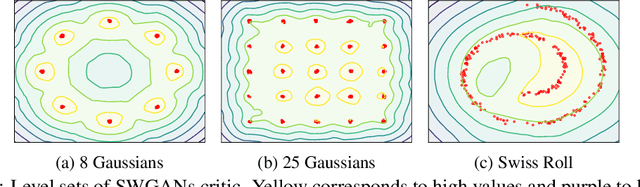
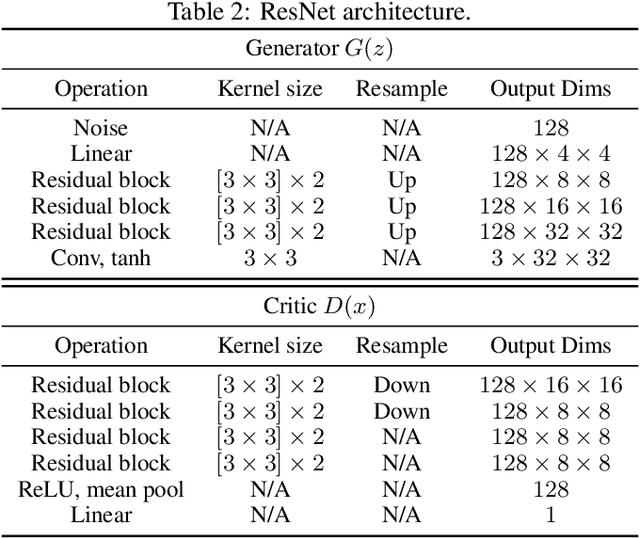
Wasserstein GANs (WGANs), built upon the Kantorovich-Rubinstein (KR) duality of Wasserstein distance, is one of the most theoretically sound GAN models. However, in practice it does not always outperform other variants of GANs. This is mostly due to the imperfect implementation of the Lipschitz condition required by the KR duality. Extensive work has been done in the community with different implementations of the Lipschitz constraint, which, however, is still hard to satisfy the restriction perfectly in practice. In this paper, we argue that the strong Lipschitz constraint might be unnecessary for optimization. Instead, we take a step back and try to relax the Lipschitz constraint. Theoretically, we first demonstrate a more general dual form of the Wasserstein distance called the Sobolev duality, which relaxes the Lipschitz constraint but still maintains the favorable gradient property of the Wasserstein distance. Moreover, we show that the KR duality is actually a special case of the Sobolev duality. Based on the relaxed duality, we further propose a generalized WGAN training scheme named Sobolev Wasserstein GAN (SWGAN), and empirically demonstrate the improvement of SWGAN over existing methods with extensive experiments.
Non-autoregressive electron flow generation for reaction prediction
Dec 16, 2020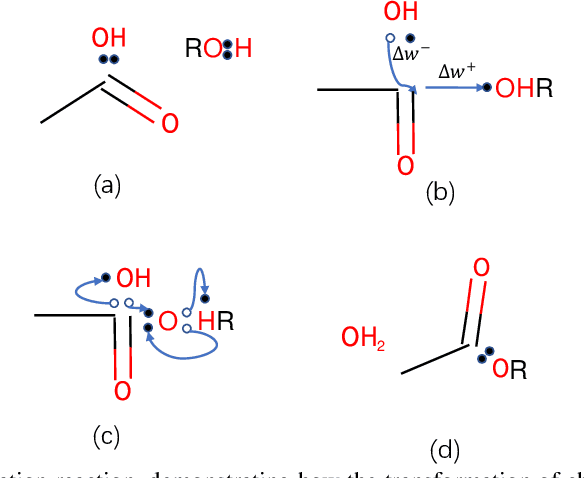
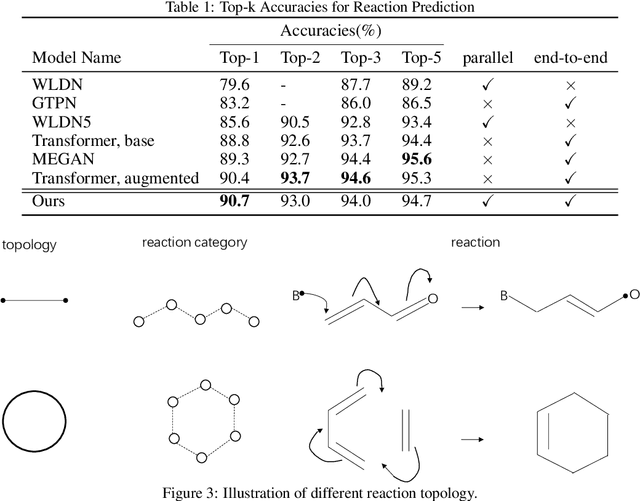
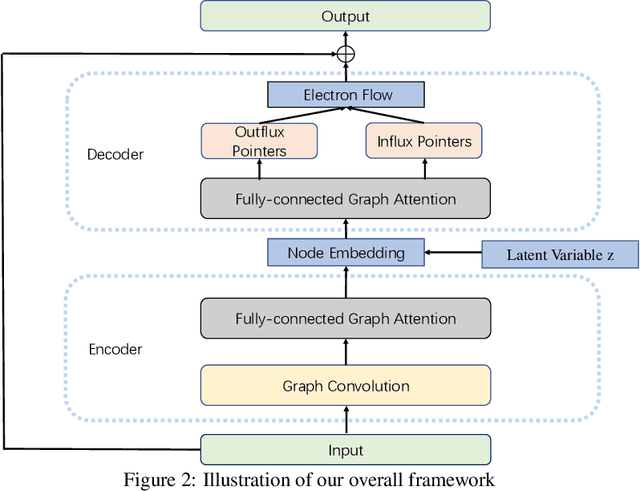

Reaction prediction is a fundamental problem in computational chemistry. Existing approaches typically generate a chemical reaction by sampling tokens or graph edits sequentially, conditioning on previously generated outputs. These autoregressive generating methods impose an arbitrary ordering of outputs and prevent parallel decoding during inference. We devise a novel decoder that avoids such sequential generating and predicts the reaction in a Non-Autoregressive manner. Inspired by physical-chemistry insights, we represent edge edits in a molecule graph as electron flows, which can then be predicted in parallel. To capture the uncertainty of reactions, we introduce latent variables to generate multi-modal outputs. Following previous works, we evaluate our model on USPTO MIT dataset. Our model achieves both an order of magnitude lower inference latency, with state-of-the-art top-1 accuracy and comparable performance on Top-K sampling.