 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe 1st PortraitCraft Challenge: A CVPR 2026 Workshop Competition on Portrait Composition Understanding and Generation
Jun 09, 2026This paper presents an overview of the inaugural PortraitCraft Challenge, held as one of the official competitions at CVPR 2026. The challenge focuses on portrait composition understanding and generation, aiming to advance AI research in portrait aesthetics analysis and controllable image synthesis. Unlike existing datasets and tasks that primarily focus on global aesthetic scoring, PortraitCraft introduces a unified evaluation framework comprising two complementary tracks. Track 1 requires models to perform structured portrait composition understanding, and Track 2 requires models to generate portrait images from structured composition descriptions under explicit compositional constraints. To support the challenge, we constructed and publicly released a large-scale portrait composition dataset consisting of approximately 50,000 curated real portrait images, providing multi-level supervision. This report describes the challenge setup, evaluation protocols, dataset composition, and final results, along with an analysis of the technical characteristics of the submitted solutions. The PortraitCraft Challenge provides a standardized and reproducible platform for research on portrait composition understanding and generation, and is expected to foster further progress in the fields of portrait aesthetics and controllable image generation.
Point Cloud Registration for Fusion between SPECT MPI and CTA Images
Apr 27, 2026Clinical fusion of Single Photon Emission Computed Tomography Myocardial Perfusion Imaging (SPECT MPI) and Computed Tomography Angiography (CTA) remains limited by cross-modality misregistration and reliance on manual landmarks, which can hinder accurate ischemia localization and lesion-level functional assessment. To address this issue, we propose a registration and fusion framework for SPECT MPI and CTA that integrates functional and structural information for comprehensive cardiac evaluation. The proposed pipeline performs U-Net-based segmentation on both modalities. On SPECT MPI, only the left ventricle (LV) is extracted, and anatomical landmarks are automatically derived from characteristic LV structures. On CTA, both ventricles are segmented, and their spatial relationship is used to automatically define landmarks at the interventricular septal junction. Scale-space consistency preprocessing and landmark-driven coarse registration are applied to mitigate initial misalignment. Based on this initialization, multiple fine registration methods are evaluated on LV epicardial surface point clouds, including ICP, SICP, CPD, CluReg, FFD, and BCPD-plus-plus. The resulting transformations are then propagated to voxel-level resampling for high-precision SPECT-CTA fusion. In a retrospective cohort of 60 patients, the proposed framework preserved sub-millimeter coronary detail from CTA while accurately overlaying quantitative SPECT perfusion. Among the evaluated methods, BCPD-plus-plus achieved the highest accuracy with a mean point cloud distance of 1.7 mm. By combining robust initialization, comparative fine registration, and voxel-level fusion, the proposed approach provides a practical solution for myocardial ischemia localization and functional evaluation of coronary lesions, while remaining independent of any specific fine registration algorithm.
CAGenMol: Condition-Aware Diffusion Language Model for Goal-Directed Molecular Generation
Apr 13, 2026Goal-directed molecular generation requires satisfying heterogeneous constraints such as protein--ligand compatibility and multi-objective drug-like properties, yet existing methods often optimize these constraints in isolation, failing to reconcile conflicting objectives (e.g., affinity vs. safety), and struggle to navigate the non-differentiable chemical space without compromising structural validity. To address these challenges, we propose CAGenMol, a condition-aware discrete diffusion framework over molecular sequences that formulates molecular design as conditional denoising guided by heterogeneous structural and property signals. By coupling discrete diffusion with reinforcement learning, the model aligns the generation trajectory with non-differentiable objectives while preserving chemical validity and diversity. The non-autoregressive nature of diffusion language model further enables iterative refinement of molecular fragments at inference time. Experiments on structure-conditioned, property-conditioned, and dual-conditioned benchmarks demonstrate consistent improvements over state-of-the-art methods in binding affinity, drug-likeness, and success rate, highlighting the effectiveness of our framework.
UGD-IML: A Unified Generative Diffusion-based Framework for Constrained and Unconstrained Image Manipulation Localization
Aug 08, 2025In the digital age, advanced image editing tools pose a serious threat to the integrity of visual content, making image forgery detection and localization a key research focus. Most existing Image Manipulation Localization (IML) methods rely on discriminative learning and require large, high-quality annotated datasets. However, current datasets lack sufficient scale and diversity, limiting model performance in real-world scenarios. To overcome this, recent studies have explored Constrained IML (CIML), which generates pixel-level annotations through algorithmic supervision. However, existing CIML approaches often depend on complex multi-stage pipelines, making the annotation process inefficient. In this work, we propose a novel generative framework based on diffusion models, named UGD-IML, which for the first time unifies both IML and CIML tasks within a single framework. By learning the underlying data distribution, generative diffusion models inherently reduce the reliance on large-scale labeled datasets, allowing our approach to perform effectively even under limited data conditions. In addition, by leveraging a class embedding mechanism and a parameter-sharing design, our model seamlessly switches between IML and CIML modes without extra components or training overhead. Furthermore, the end-to-end design enables our model to avoid cumbersome steps in the data annotation process. Extensive experimental results on multiple datasets demonstrate that UGD-IML outperforms the SOTA methods by an average of 9.66 and 4.36 in terms of F1 metrics for IML and CIML tasks, respectively. Moreover, the proposed method also excels in uncertainty estimation, visualization and robustness.
Q-CLIP: Unleashing the Power of Vision-Language Models for Video Quality Assessment through Unified Cross-Modal Adaptation
Aug 08, 2025Accurate and efficient Video Quality Assessment (VQA) has long been a key research challenge. Current mainstream VQA methods typically improve performance by pretraining on large-scale classification datasets (e.g., ImageNet, Kinetics-400), followed by fine-tuning on VQA datasets. However, this strategy presents two significant challenges: (1) merely transferring semantic knowledge learned from pretraining is insufficient for VQA, as video quality depends on multiple factors (e.g., semantics, distortion, motion, aesthetics); (2) pretraining on large-scale datasets demands enormous computational resources, often dozens or even hundreds of times greater than training directly on VQA datasets. Recently, Vision-Language Models (VLMs) have shown remarkable generalization capabilities across a wide range of visual tasks, and have begun to demonstrate promising potential in quality assessment. In this work, we propose Q-CLIP, the first fully VLMs-based framework for VQA. Q-CLIP enhances both visual and textual representations through a Shared Cross-Modal Adapter (SCMA), which contains only a minimal number of trainable parameters and is the only component that requires training. This design significantly reduces computational cost. In addition, we introduce a set of five learnable quality-level prompts to guide the VLMs in perceiving subtle quality variations, thereby further enhancing the model's sensitivity to video quality. Furthermore, we investigate the impact of different frame sampling strategies on VQA performance, and find that frame-difference-based sampling leads to better generalization performance across datasets. Extensive experiments demonstrate that Q-CLIP exhibits excellent performance on several VQA datasets.
DS-ProGen: A Dual-Structure Deep Language Model for Functional Protein Design
May 18, 2025Inverse Protein Folding (IPF) is a critical subtask in the field of protein design, aiming to engineer amino acid sequences capable of folding correctly into a specified three-dimensional (3D) conformation. Although substantial progress has been achieved in recent years, existing methods generally rely on either backbone coordinates or molecular surface features alone, which restricts their ability to fully capture the complex chemical and geometric constraints necessary for precise sequence prediction. To address this limitation, we present DS-ProGen, a dual-structure deep language model for functional protein design, which integrates both backbone geometry and surface-level representations. By incorporating backbone coordinates as well as surface chemical and geometric descriptors into a next-amino-acid prediction paradigm, DS-ProGen is able to generate functionally relevant and structurally stable sequences while satisfying both global and local conformational constraints. On the PRIDE dataset, DS-ProGen attains the current state-of-the-art recovery rate of 61.47%, demonstrating the synergistic advantage of multi-modal structural encoding in protein design. Furthermore, DS-ProGen excels in predicting interactions with a variety of biological partners, including ligands, ions, and RNA, confirming its robust functional retention capabilities.
UPMAD-Net: A Brain Tumor Segmentation Network with Uncertainty Guidance and Adaptive Multimodal Feature Fusion
May 06, 2025
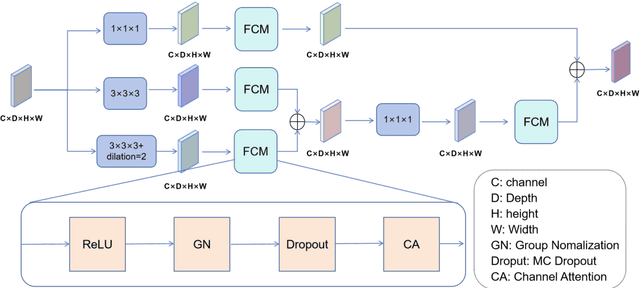


Background: Brain tumor segmentation has a significant impact on the diagnosis and treatment of brain tumors. Accurate brain tumor segmentation remains challenging due to their irregular shapes, vague boundaries, and high variability. Objective: We propose a brain tumor segmentation method that combines deep learning with prior knowledge derived from a region-growing algorithm. Methods: The proposed method utilizes a multi-scale feature fusion (MSFF) module and adaptive attention mechanisms (AAM) to extract multi-scale features and capture global contextual information. To enhance the model's robustness in low-confidence regions, the Monte Carlo Dropout (MC Dropout) strategy is employed for uncertainty estimation. Results: Extensive experiments demonstrate that the proposed method achieves superior performance on Brain Tumor Segmentation (BraTS) datasets, significantly outperforming various state-of-the-art methods. On the BraTS2021 dataset, the test Dice scores are 89.18% for Enhancing Tumor (ET) segmentation, 93.67% for Whole Tumor (WT) segmentation, and 91.23% for Tumor Core (TC) segmentation. On the BraTS2019 validation set, the validation Dice scores are 87.43%, 90.92%, and 90.40% for ET, WT, and TC segmentation, respectively. Ablation studies further confirmed the contribution of each module to segmentation accuracy, indicating that each component played a vital role in overall performance improvement. Conclusion: This study proposed a novel 3D brain tumor segmentation network based on the U-Net architecture. By incorporating the prior knowledge and employing the uncertainty estimation method, the robustness and performance were improved. The code for the proposed method is available at https://github.com/chenzhao2023/UPMAD_Net_BrainSeg.
Myocardial Region-guided Feature Aggregation Net for Automatic Coronary artery Segmentation and Stenosis Assessment using Coronary Computed Tomography Angiography
Apr 27, 2025



Coronary artery disease (CAD) remains a leading cause of mortality worldwide, requiring accurate segmentation and stenosis detection using Coronary Computed Tomography angiography (CCTA). Existing methods struggle with challenges such as low contrast, morphological variability and small vessel segmentation. To address these limitations, we propose the Myocardial Region-guided Feature Aggregation Net, a novel U-shaped dual-encoder architecture that integrates anatomical prior knowledge to enhance robustness in coronary artery segmentation. Our framework incorporates three key innovations: (1) a Myocardial Region-guided Module that directs attention to coronary regions via myocardial contour expansion and multi-scale feature fusion, (2) a Residual Feature Extraction Encoding Module that combines parallel spatial channel attention with residual blocks to enhance local-global feature discrimination, and (3) a Multi-scale Feature Fusion Module for adaptive aggregation of hierarchical vascular features. Additionally, Monte Carlo dropout f quantifies prediction uncertainty, supporting clinical interpretability. For stenosis detection, a morphology-based centerline extraction algorithm separates the vascular tree into anatomical branches, enabling cross-sectional area quantification and stenosis grading. The superiority of MGFA-Net was demonstrated by achieving an Dice score of 85.04%, an accuracy of 84.24%, an HD95 of 6.1294 mm, and an improvement of 5.46% in true positive rate for stenosis detection compared to3D U-Net. The integrated segmentation-to-stenosis pipeline provides automated, clinically interpretable CAD assessment, bridging deep learning with anatomical prior knowledge for precision medicine. Our code is publicly available at http://github.com/chenzhao2023/MGFA_CCTA
UniGenX: Unified Generation of Sequence and Structure with Autoregressive Diffusion
Mar 09, 2025Unified generation of sequence and structure for scientific data (e.g., materials, molecules, proteins) is a critical task. Existing approaches primarily rely on either autoregressive sequence models or diffusion models, each offering distinct advantages and facing notable limitations. Autoregressive models, such as GPT, Llama, and Phi-4, have demonstrated remarkable success in natural language generation and have been extended to multimodal tasks (e.g., image, video, and audio) using advanced encoders like VQ-VAE to represent complex modalities as discrete sequences. However, their direct application to scientific domains is challenging due to the high precision requirements and the diverse nature of scientific data. On the other hand, diffusion models excel at generating high-dimensional scientific data, such as protein, molecule, and material structures, with remarkable accuracy. Yet, their inability to effectively model sequences limits their potential as general-purpose multimodal foundation models. To address these challenges, we propose UniGenX, a unified framework that combines autoregressive next-token prediction with conditional diffusion models. This integration leverages the strengths of autoregressive models to ease the training of conditional diffusion models, while diffusion-based generative heads enhance the precision of autoregressive predictions. We validate the effectiveness of UniGenX on material and small molecule generation tasks, achieving a significant leap in state-of-the-art performance for material crystal structure prediction and establishing new state-of-the-art results for small molecule structure prediction, de novo design, and conditional generation. Notably, UniGenX demonstrates significant improvements, especially in handling long sequences for complex structures, showcasing its efficacy as a versatile tool for scientific data generation.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.