 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeArtificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023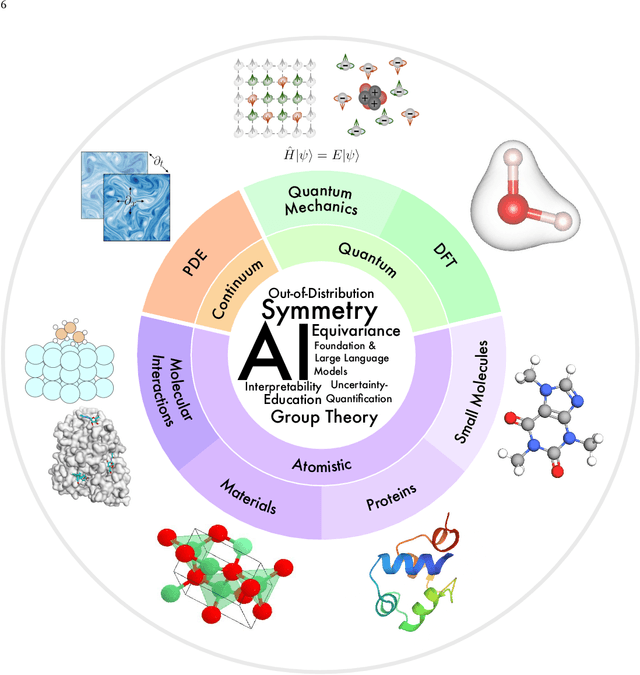



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Optimizing protein fitness using Gibbs sampling with Graph-based Smoothing
Jul 02, 2023



The ability to design novel proteins with higher fitness on a given task would be revolutionary for many fields of medicine. However, brute-force search through the combinatorially large space of sequences is infeasible. Prior methods constrain search to a small mutational radius from a reference sequence, but such heuristics drastically limit the design space. Our work seeks to remove the restriction on mutational distance while enabling efficient exploration. We propose Gibbs sampling with Graph-based Smoothing (GGS) which iteratively applies Gibbs with gradients to propose advantageous mutations using graph-based smoothing to remove noisy gradients that lead to false positives. Our method is state-of-the-art in discovering high-fitness proteins with up to 8 mutations from the training set. We study the GFP and AAV design problems, ablations, and baselines to elucidate the results. Code: https://github.com/kirjner/GGS
Restart Sampling for Improving Generative Processes
Jun 26, 2023



Generative processes that involve solving differential equations, such as diffusion models, frequently necessitate balancing speed and quality. ODE-based samplers are fast but plateau in performance while SDE-based samplers deliver higher sample quality at the cost of increased sampling time. We attribute this difference to sampling errors: ODE-samplers involve smaller discretization errors while stochasticity in SDE contracts accumulated errors. Based on these findings, we propose a novel sampling algorithm called Restart in order to better balance discretization errors and contraction. The sampling method alternates between adding substantial noise in additional forward steps and strictly following a backward ODE. Empirically, Restart sampler surpasses previous SDE and ODE samplers in both speed and accuracy. Restart not only outperforms the previous best SDE results, but also accelerates the sampling speed by 10-fold / 2-fold on CIFAR-10 / ImageNet $64 \times 64$. In addition, it attains significantly better sample quality than ODE samplers within comparable sampling times. Moreover, Restart better balances text-image alignment/visual quality versus diversity than previous samplers in the large-scale text-to-image Stable Diffusion model pre-trained on LAION $512 \times 512$. Code is available at https://github.com/Newbeeer/diffusion_restart_sampling
Towards Coherent Image Inpainting Using Denoising Diffusion Implicit Models
Apr 06, 2023Image inpainting refers to the task of generating a complete, natural image based on a partially revealed reference image. Recently, many research interests have been focused on addressing this problem using fixed diffusion models. These approaches typically directly replace the revealed region of the intermediate or final generated images with that of the reference image or its variants. However, since the unrevealed regions are not directly modified to match the context, it results in incoherence between revealed and unrevealed regions. To address the incoherence problem, a small number of methods introduce a rigorous Bayesian framework, but they tend to introduce mismatches between the generated and the reference images due to the approximation errors in computing the posterior distributions. In this paper, we propose COPAINT, which can coherently inpaint the whole image without introducing mismatches. COPAINT also uses the Bayesian framework to jointly modify both revealed and unrevealed regions, but approximates the posterior distribution in a way that allows the errors to gradually drop to zero throughout the denoising steps, thus strongly penalizing any mismatches with the reference image. Our experiments verify that COPAINT can outperform the existing diffusion-based methods under both objective and subjective metrics. The codes are available at https://github.com/UCSB-NLP-Chang/CoPaint/.
GenPhys: From Physical Processes to Generative Models
Apr 05, 2023



Since diffusion models (DM) and the more recent Poisson flow generative models (PFGM) are inspired by physical processes, it is reasonable to ask: Can physical processes offer additional new generative models? We show that the answer is yes. We introduce a general family, Generative Models from Physical Processes (GenPhys), where we translate partial differential equations (PDEs) describing physical processes to generative models. We show that generative models can be constructed from s-generative PDEs (s for smooth). GenPhys subsume the two existing generative models (DM and PFGM) and even give rise to new families of generative models, e.g., "Yukawa Generative Models" inspired from weak interactions. On the other hand, some physical processes by default do not belong to the GenPhys family, e.g., the wave equation and the Schr\"{o}dinger equation, but could be made into the GenPhys family with some modifications. Our goal with GenPhys is to explore and expand the design space of generative models.
EigenFold: Generative Protein Structure Prediction with Diffusion Models
Apr 05, 2023



Protein structure prediction has reached revolutionary levels of accuracy on single structures, yet distributional modeling paradigms are needed to capture the conformational ensembles and flexibility that underlie biological function. Towards this goal, we develop EigenFold, a diffusion generative modeling framework for sampling a distribution of structures from a given protein sequence. We define a diffusion process that models the structure as a system of harmonic oscillators and which naturally induces a cascading-resolution generative process along the eigenmodes of the system. On recent CAMEO targets, EigenFold achieves a median TMScore of 0.84, while providing a more comprehensive picture of model uncertainty via the ensemble of sampled structures relative to existing methods. We then assess EigenFold's ability to model and predict conformational heterogeneity for fold-switching proteins and ligand-induced conformational change. Code is available at https://github.com/bjing2016/EigenFold.
Stable Target Field for Reduced Variance Score Estimation in Diffusion Models
Feb 17, 2023



Diffusion models generate samples by reversing a fixed forward diffusion process. Despite already providing impressive empirical results, these diffusion models algorithms can be further improved by reducing the variance of the training targets in their denoising score-matching objective. We argue that the source of such variance lies in the handling of intermediate noise-variance scales, where multiple modes in the data affect the direction of reverse paths. We propose to remedy the problem by incorporating a reference batch which we use to calculate weighted conditional scores as more stable training targets. We show that the procedure indeed helps in the challenging intermediate regime by reducing (the trace of) the covariance of training targets. The new stable targets can be seen as trading bias for reduced variance, where the bias vanishes with increasing reference batch size. Empirically, we show that the new objective improves the image quality, stability, and training speed of various popular diffusion models across datasets with both general ODE and SDE solvers. When used in combination with EDM, our method yields a current SOTA FID of 1.90 with 35 network evaluations on the unconditional CIFAR-10 generation task. The code is available at https://github.com/Newbeeer/stf
SE(3) diffusion model with application to protein backbone generation
Feb 11, 2023



The design of novel protein structures remains a challenge in protein engineering for applications across biomedicine and chemistry. In this line of work, a diffusion model over rigid bodies in 3D (referred to as frames) has shown success in generating novel, functional protein backbones that have not been observed in nature. However, there exists no principled methodological framework for diffusion on SE(3), the space of orientation preserving rigid motions in R3, that operates on frames and confers the group invariance. We address these shortcomings by developing theoretical foundations of SE(3) invariant diffusion models on multiple frames followed by a novel framework, FrameDiff, for learning the SE(3) equivariant score over multiple frames. We apply FrameDiff on monomer backbone generation and find it can generate designable monomers up to 500 amino acids without relying on a pretrained protein structure prediction network that has been integral to previous methods. We find our samples are capable of generalizing beyond any known protein structure.
PFGM++: Unlocking the Potential of Physics-Inspired Generative Models
Feb 10, 2023We introduce a new family of physics-inspired generative models termed PFGM++ that unifies diffusion models and Poisson Flow Generative Models (PFGM). These models realize generative trajectories for $N$ dimensional data by embedding paths in $N{+}D$ dimensional space while still controlling the progression with a simple scalar norm of the $D$ additional variables. The new models reduce to PFGM when $D{=}1$ and to diffusion models when $D{\to}\infty$. The flexibility of choosing $D$ allows us to trade off robustness against rigidity as increasing $D$ results in more concentrated coupling between the data and the additional variable norms. We dispense with the biased large batch field targets used in PFGM and instead provide an unbiased perturbation-based objective similar to diffusion models. To explore different choices of $D$, we provide a direct alignment method for transferring well-tuned hyperparameters from diffusion models ($D{\to} \infty$) to any finite $D$ values. Our experiments show that models with finite $D$ can be superior to previous state-of-the-art diffusion models on CIFAR-10/FFHQ $64{\times}64$ datasets, with FID scores of $1.91/2.43$ when $D{=}2048/128$. In class-conditional setting, $D{=}2048$ yields current state-of-the-art FID of $1.74$ on CIFAR-10. In addition, we demonstrate that models with smaller $D$ exhibit improved robustness against modeling errors. Code is available at https://github.com/Newbeeer/pfgmpp
Is Conditional Generative Modeling all you need for Decision-Making?
Dec 07, 2022



Recent improvements in conditional generative modeling have made it possible to generate high-quality images from language descriptions alone. We investigate whether these methods can directly address the problem of sequential decision-making. We view decision-making not through the lens of reinforcement learning (RL), but rather through conditional generative modeling. To our surprise, we find that our formulation leads to policies that can outperform existing offline RL approaches across standard benchmarks. By modeling a policy as a return-conditional diffusion model, we illustrate how we may circumvent the need for dynamic programming and subsequently eliminate many of the complexities that come with traditional offline RL. We further demonstrate the advantages of modeling policies as conditional diffusion models by considering two other conditioning variables: constraints and skills. Conditioning on a single constraint or skill during training leads to behaviors at test-time that can satisfy several constraints together or demonstrate a composition of skills. Our results illustrate that conditional generative modeling is a powerful tool for decision-making.