 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHandling Missing Data via Max-Entropy Regularized Graph Autoencoder
Nov 30, 2022



Graph neural networks (GNNs) are popular weapons for modeling relational data. Existing GNNs are not specified for attribute-incomplete graphs, making missing attribute imputation a burning issue. Until recently, many works notice that GNNs are coupled with spectral concentration, which means the spectrum obtained by GNNs concentrates on a local part in spectral domain, e.g., low-frequency due to oversmoothing issue. As a consequence, GNNs may be seriously flawed for reconstructing graph attributes as graph spectral concentration tends to cause a low imputation precision. In this work, we present a regularized graph autoencoder for graph attribute imputation, named MEGAE, which aims at mitigating spectral concentration problem by maximizing the graph spectral entropy. Notably, we first present the method for estimating graph spectral entropy without the eigen-decomposition of Laplacian matrix and provide the theoretical upper error bound. A maximum entropy regularization then acts in the latent space, which directly increases the graph spectral entropy. Extensive experiments show that MEGAE outperforms all the other state-of-the-art imputation methods on a variety of benchmark datasets.
Towards Complete-View and High-Level Pose-based Gait Recognition
Sep 23, 2022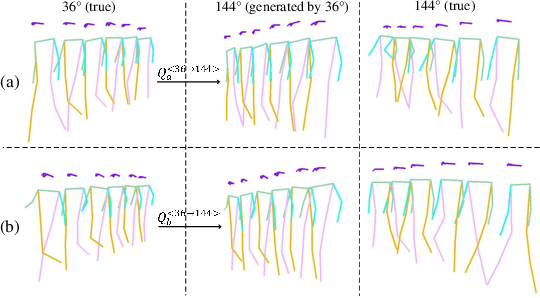
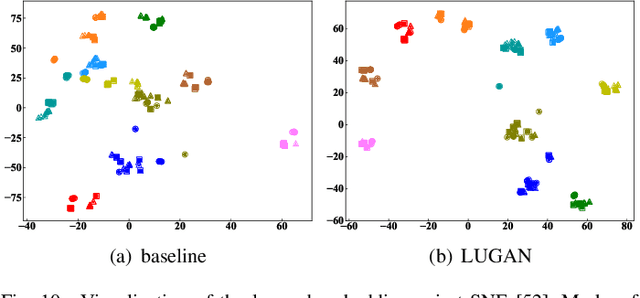
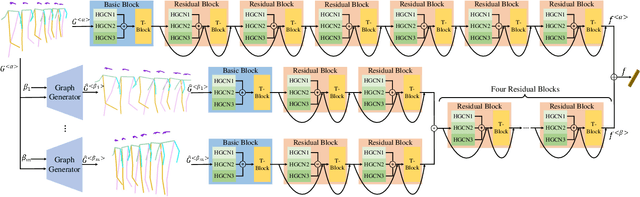
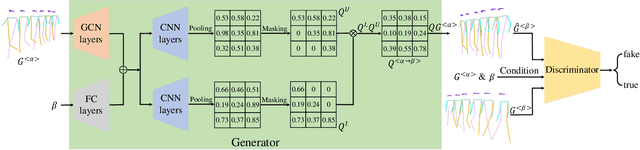
The model-based gait recognition methods usually adopt the pedestrian walking postures to identify human beings. However, existing methods did not explicitly resolve the large intra-class variance of human pose due to camera views changing. In this paper, we propose to generate multi-view pose sequences for each single-view pose sample by learning full-rank transformation matrices via lower-upper generative adversarial network (LUGAN). By the prior of camera imaging, we derive that the spatial coordinates between cross-view poses satisfy a linear transformation of a full-rank matrix, thereby, this paper employs the adversarial training to learn transformation matrices from the source pose and target views to obtain the target pose sequences. To this end, we implement a generator composed of graph convolutional (GCN) layers, fully connected (FC) layers and two-branch convolutional (CNN) layers: GCN layers and FC layers encode the source pose sequence and target view, then CNN branches learn a lower triangular matrix and an upper triangular matrix, respectively, finally they are multiplied to formulate the full-rank transformation matrix. For the purpose of adversarial training, we further devise a condition discriminator that distinguishes whether the pose sequence is true or generated. To enable the high-level correlation learning, we propose a plug-and-play module, named multi-scale hypergraph convolution (HGC), to replace the spatial graph convolutional layer in baseline, which could simultaneously model the joint-level, part-level and body-level correlations. Extensive experiments on two large gait recognition datasets, i.e., CASIA-B and OUMVLP-Pose, demonstrate that our method outperforms the baseline model and existing pose-based methods by a large margin.
MDM: Molecular Diffusion Model for 3D Molecule Generation
Sep 13, 2022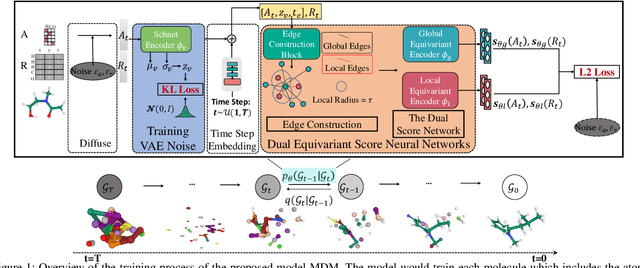
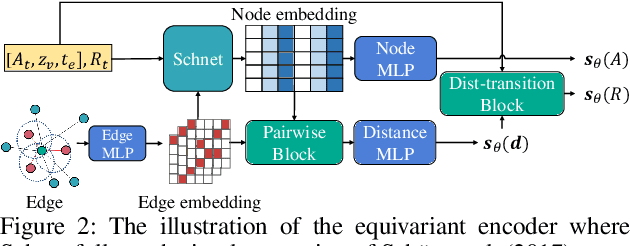
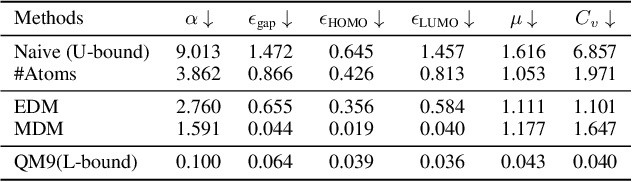
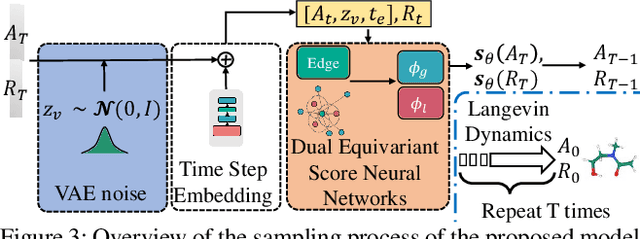
Molecule generation, especially generating 3D molecular geometries from scratch (i.e., 3D \textit{de novo} generation), has become a fundamental task in drug designs. Existing diffusion-based 3D molecule generation methods could suffer from unsatisfactory performances, especially when generating large molecules. At the same time, the generated molecules lack enough diversity. This paper proposes a novel diffusion model to address those two challenges. First, interatomic relations are not in molecules' 3D point cloud representations. Thus, it is difficult for existing generative models to capture the potential interatomic forces and abundant local constraints. To tackle this challenge, we propose to augment the potential interatomic forces and further involve dual equivariant encoders to encode interatomic forces of different strengths. Second, existing diffusion-based models essentially shift elements in geometry along the gradient of data density. Such a process lacks enough exploration in the intermediate steps of the Langevin dynamics. To address this issue, we introduce a distributional controlling variable in each diffusion/reverse step to enforce thorough explorations and further improve generation diversity. Extensive experiments on multiple benchmarks demonstrate that the proposed model significantly outperforms existing methods for both unconditional and conditional generation tasks. We also conduct case studies to help understand the physicochemical properties of the generated molecules.
Similarity-aware Positive Instance Sampling for Graph Contrastive Pre-training
Jun 23, 2022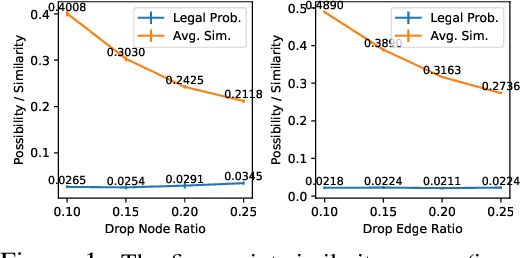
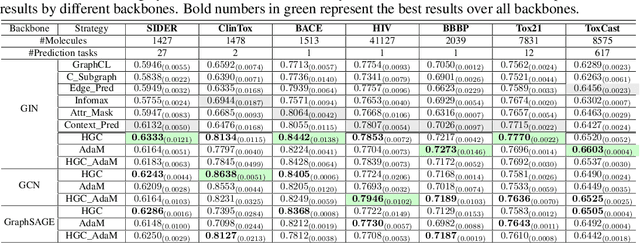
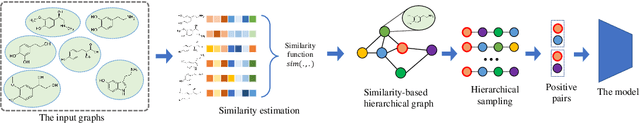
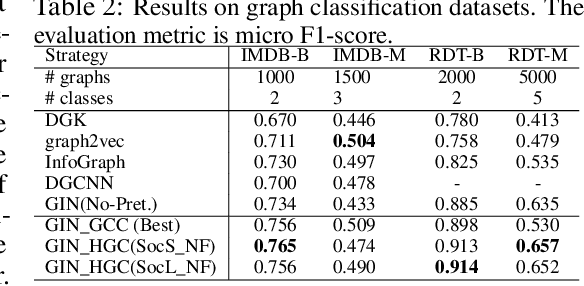
Graph instance contrastive learning has been proved as an effective task for Graph Neural Network (GNN) pre-training. However, one key issue may seriously impede the representative power in existing works: Positive instances created by current methods often miss crucial information of graphs or even yield illegal instances (such as non-chemically-aware graphs in molecular generation). To remedy this issue, we propose to select positive graph instances directly from existing graphs in the training set, which ultimately maintains the legality and similarity to the target graphs. Our selection is based on certain domain-specific pair-wise similarity measurements as well as sampling from a hierarchical graph encoding similarity relations among graphs. Besides, we develop an adaptive node-level pre-training method to dynamically mask nodes to distribute them evenly in the graph. We conduct extensive experiments on $13$ graph classification and node classification benchmark datasets from various domains. The results demonstrate that the GNN models pre-trained by our strategies can outperform those trained-from-scratch models as well as the variants obtained by existing methods.
Tyger: Task-Type-Generic Active Learning for Molecular Property Prediction
May 23, 2022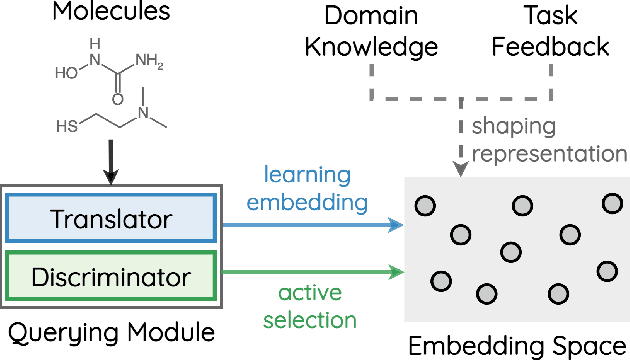
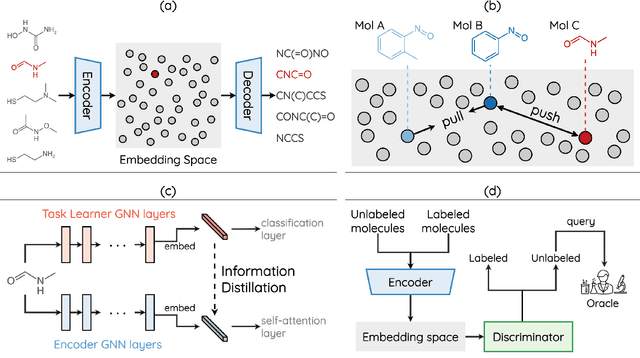
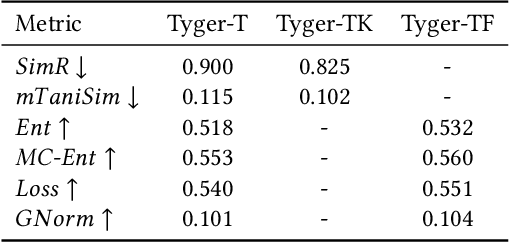
How to accurately predict the properties of molecules is an essential problem in AI-driven drug discovery, which generally requires a large amount of annotation for training deep learning models. Annotating molecules, however, is quite costly because it requires lab experiments conducted by experts. To reduce annotation cost, deep Active Learning (AL) methods are developed to select only the most representative and informative data for annotating. However, existing best deep AL methods are mostly developed for a single type of learning task (e.g., single-label classification), and hence may not perform well in molecular property prediction that involves various task types. In this paper, we propose a Task-type-generic active learning framework (termed Tyger) that is able to handle different types of learning tasks in a unified manner. The key is to learn a chemically-meaningful embedding space and perform active selection fully based on the embeddings, instead of relying on task-type-specific heuristics (e.g., class-wise prediction probability) as done in existing works. Specifically, for learning the embedding space, we instantiate a querying module that learns to translate molecule graphs into corresponding SMILES strings. Furthermore, to ensure that samples selected from the space are both representative and informative, we propose to shape the embedding space by two learning objectives, one based on domain knowledge and the other leveraging feedback from the task learner (i.e., model that performs the learning task at hand). We conduct extensive experiments on benchmark datasets of different task types. Experimental results show that Tyger consistently achieves high AL performance on molecular property prediction, outperforming baselines by a large margin. We also perform ablative experiments to verify the effectiveness of each component in Tyger.
A Survey of Trustworthy Graph Learning: Reliability, Explainability, and Privacy Protection
May 23, 2022

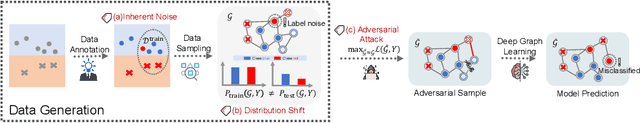
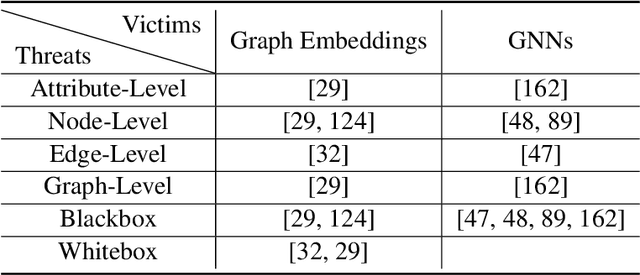
Deep graph learning has achieved remarkable progresses in both business and scientific areas ranging from finance and e-commerce, to drug and advanced material discovery. Despite these progresses, how to ensure various deep graph learning algorithms behave in a socially responsible manner and meet regulatory compliance requirements becomes an emerging problem, especially in risk-sensitive domains. Trustworthy graph learning (TwGL) aims to solve the above problems from a technical viewpoint. In contrast to conventional graph learning research which mainly cares about model performance, TwGL considers various reliability and safety aspects of the graph learning framework including but not limited to robustness, explainability, and privacy. In this survey, we provide a comprehensive review of recent leading approaches in the TwGL field from three dimensions, namely, reliability, explainability, and privacy protection. We give a general categorization for existing work and review typical work for each category. To give further insights for TwGL research, we provide a unified view to inspect previous works and build the connection between them. We also point out some important open problems remaining to be solved in the future developments of TwGL.
GPN: A Joint Structural Learning Framework for Graph Neural Networks
May 12, 2022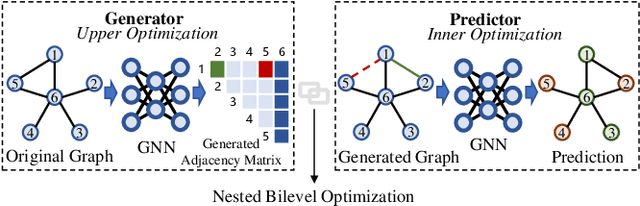
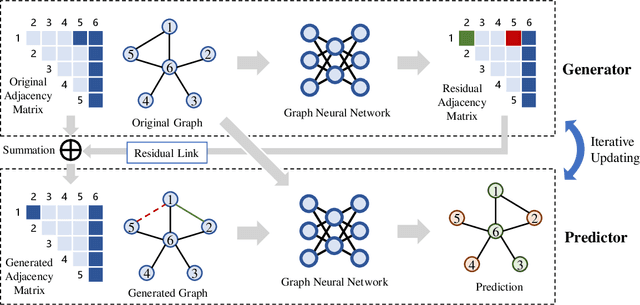
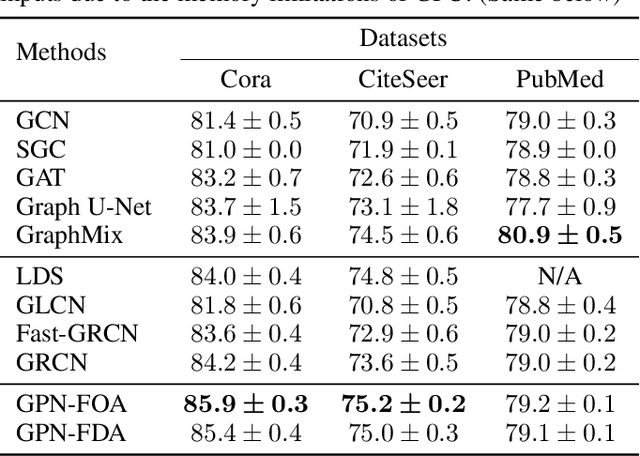
Graph neural networks (GNNs) have been applied into a variety of graph tasks. Most existing work of GNNs is based on the assumption that the given graph data is optimal, while it is inevitable that there exists missing or incomplete edges in the graph data for training, leading to degraded performance. In this paper, we propose Generative Predictive Network (GPN), a GNN-based joint learning framework that simultaneously learns the graph structure and the downstream task. Specifically, we develop a bilevel optimization framework for this joint learning task, in which the upper optimization (generator) and the lower optimization (predictor) are both instantiated with GNNs. To the best of our knowledge, our method is the first GNN-based bilevel optimization framework for resolving this task. Through extensive experiments, our method outperforms a wide range of baselines using benchmark datasets.
Hypergraph Convolutional Networks via Equivalency between Hypergraphs and Undirected Graphs
Apr 06, 2022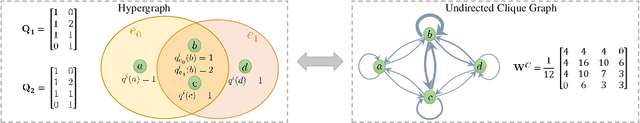
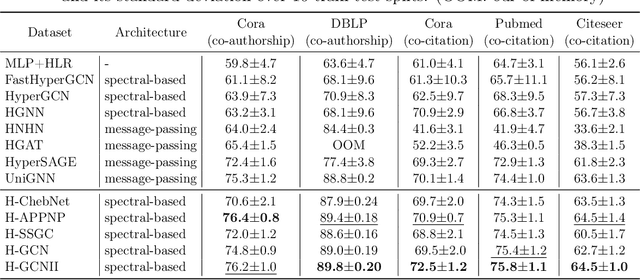
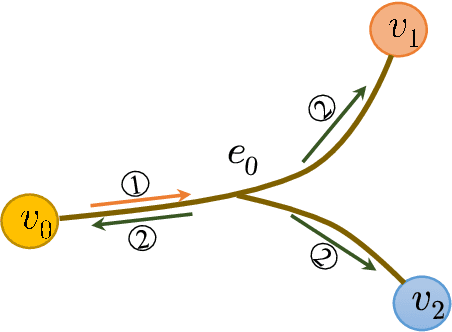
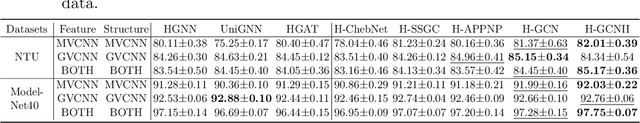
As a powerful tool for modeling complex relationships, hypergraphs are gaining popularity from the graph learning community. However, commonly used frameworks in deep hypergraph learning focus on hypergraphs with \textit{edge-independent vertex weights}(EIVWs), without considering hypergraphs with \textit{edge-dependent vertex weights} (EDVWs) that have more modeling power. To compensate for this, in this paper, we present General Hypergraph Spectral Convolution(GHSC), a general learning framework that not only can handle EDVW and EIVW hypergraphs, but more importantly, enables theoretically explicitly utilizing the existing powerful Graph Convolutional Neural Networks (GCNNs) such that largely ease the design of Hypergraph Neural Networks. In this framework, the graph Laplacian of the given undirected GCNNs is replaced with a unified hypergraph Laplacian that incorporates vertex weight information from a random walk perspective by equating our defined generalized hypergraphs with simple undirected graphs. Extensive experiments from various domains including social network analysis, visual objective classification, protein learning demonstrate that the proposed framework can achieve state-of-the-art performance.
Smoothing Matters: Momentum Transformer for Domain Adaptive Semantic Segmentation
Mar 15, 2022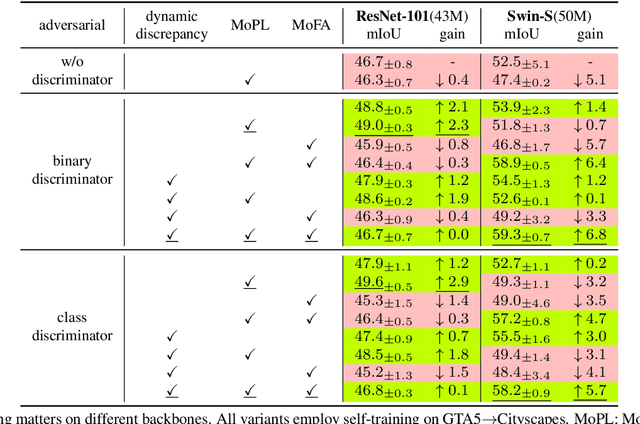
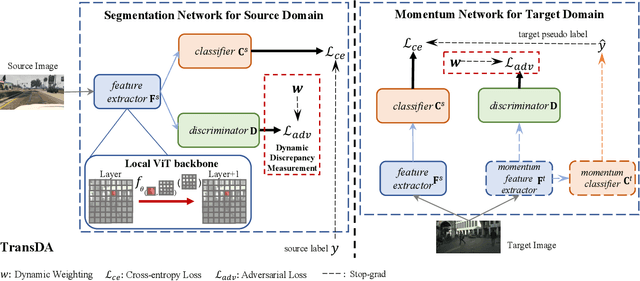

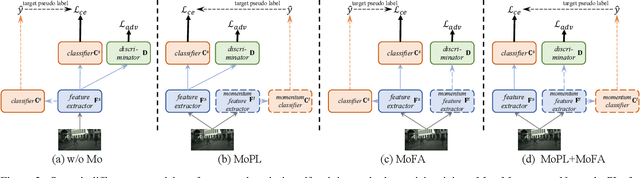
After the great success of Vision Transformer variants (ViTs) in computer vision, it has also demonstrated great potential in domain adaptive semantic segmentation. Unfortunately, straightforwardly applying local ViTs in domain adaptive semantic segmentation does not bring in expected improvement. We find that the pitfall of local ViTs is due to the severe high-frequency components generated during both the pseudo-label construction and features alignment for target domains. These high-frequency components make the training of local ViTs very unsmooth and hurt their transferability. In this paper, we introduce a low-pass filtering mechanism, momentum network, to smooth the learning dynamics of target domain features and pseudo labels. Furthermore, we propose a dynamic of discrepancy measurement to align the distributions in the source and target domains via dynamic weights to evaluate the importance of the samples. After tackling the above issues, extensive experiments on sim2real benchmarks show that the proposed method outperforms the state-of-the-art methods. Our codes are available at https://github.com/alpc91/TransDA
Equivariant Graph Mechanics Networks with Constraints
Mar 12, 2022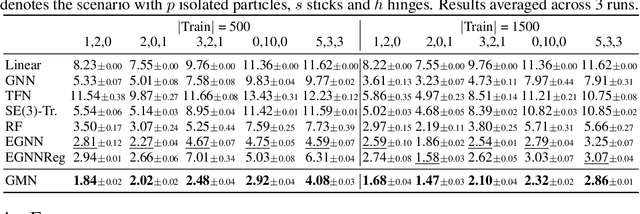
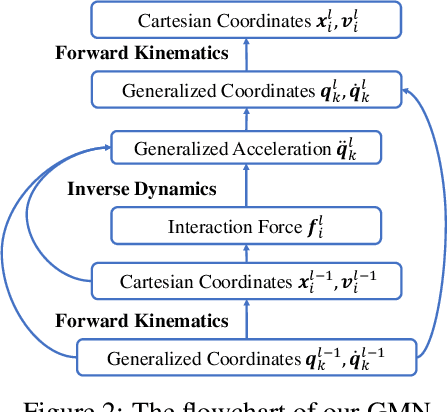
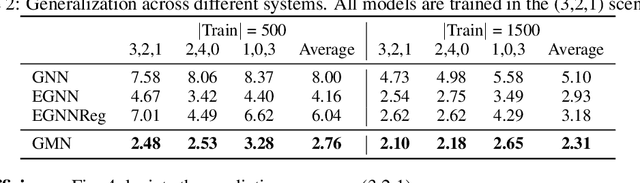
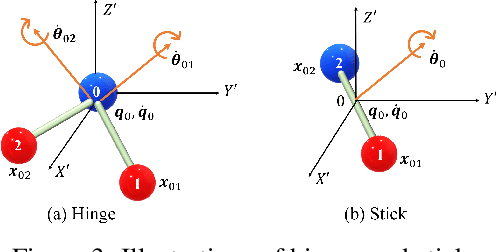
Learning to reason about relations and dynamics over multiple interacting objects is a challenging topic in machine learning. The challenges mainly stem from that the interacting systems are exponentially-compositional, symmetrical, and commonly geometrically-constrained. Current methods, particularly the ones based on equivariant Graph Neural Networks (GNNs), have targeted on the first two challenges but remain immature for constrained systems. In this paper, we propose Graph Mechanics Network (GMN) which is combinatorially efficient, equivariant and constraint-aware. The core of GMN is that it represents, by generalized coordinates, the forward kinematics information (positions and velocities) of a structural object. In this manner, the geometrical constraints are implicitly and naturally encoded in the forward kinematics. Moreover, to allow equivariant message passing in GMN, we have developed a general form of orthogonality-equivariant functions, given that the dynamics of constrained systems are more complicated than the unconstrained counterparts. Theoretically, the proposed equivariant formulation is proved to be universally expressive under certain conditions. Extensive experiments support the advantages of GMN compared to the state-of-the-art GNNs in terms of prediction accuracy, constraint satisfaction and data efficiency on the simulated systems consisting of particles, sticks and hinges, as well as two real-world datasets for molecular dynamics prediction and human motion capture.