 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMAT-Cell: A Multi-Agent Tree-Structured Reasoning Framework for Batch-Level Single-Cell Annotation
Apr 07, 2026Automated cellular reasoning faces a core dichotomy: supervised methods fall into the Reference Trap and fail to generalize to out-of-distribution cell states, while large language models (LLMs), without grounded biological priors, suffer from a Signal-to-Noise Paradox that produces spurious associations. We propose MAT-Cell, a neuro-symbolic reasoning framework that reframes single-cell analysis from black-box classification into constructive, verifiable proof generation. MAT-Cell injects symbolic constraints through adaptive Retrieval-Augmented Generation (RAG) to ground neural reasoning in biological axioms and reduce transcriptomic noise. It further employs a dialectic verification process with homogeneous rebuttal agents to audit and prune reasoning paths, forming syllogistic derivation trees that enforce logical consistency.Across large-scale and cross-species benchmarks, MAT-Cell significantly outperforms state-of-the-art (SOTA) models and maintains robust per-formance in challenging scenarios where baselinemethods severely degrade. Code is available at https://gith ub.com/jiangliu91/MAT-Cell-A-Mul ti-Agent-Tree-Structured-Reasoni ng-Framework-for-Batch-Level-Sin gle-Cell-Annotation.
Beyond the Academic Monoculture: A Unified Framework and Industrial Perspective for Attributed Graph Clustering
Mar 21, 2026Attributed Graph Clustering (AGC) is a fundamental unsupervised task that partitions nodes into cohesive groups by jointly modeling structural topology and node attributes. While the advent of graph neural networks and self-supervised learning has catalyzed a proliferation of AGC methodologies, a widening chasm persists between academic benchmark performance and the stringent demands of real-world industrial deployment. To bridge this gap, this survey provides a comprehensive, industrially grounded review of AGC from three complementary perspectives. First, we introduce the Encode-Cluster-Optimize taxonomic framework, which decomposes the diverse algorithmic landscape into three orthogonal, composable modules: representation encoding, cluster projection, and optimization strategy. This unified paradigm enables principled architectural comparisons and inspires novel methodological combinations. Second, we critically examine prevailing evaluation protocols to expose the field's academic monoculture: a pervasive over-reliance on small, homophilous citation networks, the inadequacy of supervised-only metrics for an inherently unsupervised task, and the chronic neglect of computational scalability. In response, we advocate for a holistic evaluation standard that integrates supervised semantic alignment, unsupervised structural integrity, and rigorous efficiency profiling. Third, we explicitly confront the practical realities of industrial deployment. By analyzing operational constraints such as massive scale, severe heterophily, and tabular feature noise alongside extensive empirical evidence from our companion benchmark, we outline actionable engineering strategies. Furthermore, we chart a clear roadmap for future research, prioritizing heterophily-robust encoders, scalable joint optimization, and unsupervised model selection criteria to meet production-grade requirements.
The Trinity of Consistency as a Defining Principle for General World Models
Feb 26, 2026The construction of World Models capable of learning, simulating, and reasoning about objective physical laws constitutes a foundational challenge in the pursuit of Artificial General Intelligence. Recent advancements represented by video generation models like Sora have demonstrated the potential of data-driven scaling laws to approximate physical dynamics, while the emerging Unified Multimodal Model (UMM) offers a promising architectural paradigm for integrating perception, language, and reasoning. Despite these advances, the field still lacks a principled theoretical framework that defines the essential properties requisite for a General World Model. In this paper, we propose that a World Model must be grounded in the Trinity of Consistency: Modal Consistency as the semantic interface, Spatial Consistency as the geometric basis, and Temporal Consistency as the causal engine. Through this tripartite lens, we systematically review the evolution of multimodal learning, revealing a trajectory from loosely coupled specialized modules toward unified architectures that enable the synergistic emergence of internal world simulators. To complement this conceptual framework, we introduce CoW-Bench, a benchmark centered on multi-frame reasoning and generation scenarios. CoW-Bench evaluates both video generation models and UMMs under a unified evaluation protocol. Our work establishes a principled pathway toward general world models, clarifying both the limitations of current systems and the architectural requirements for future progress.
VecFormer: Towards Efficient and Generalizable Graph Transformer with Graph Token Attention
Feb 23, 2026Graph Transformer has demonstrated impressive capabilities in the field of graph representation learning. However, existing approaches face two critical challenges: (1) most models suffer from exponentially increasing computational complexity, making it difficult to scale to large graphs; (2) attention mechanisms based on node-level operations limit the flexibility of the model and result in poor generalization performance in out-of-distribution (OOD) scenarios. To address these issues, we propose \textbf{VecFormer} (the \textbf{Vec}tor Quantized Graph Trans\textbf{former}), an efficient and highly generalizable model for node classification, particularly under OOD settings. VecFormer adopts a two-stage training paradigm. In the first stage, two codebooks are used to reconstruct the node features and the graph structure, aiming to learn the rich semantic \texttt{Graph Codes}. In the second stage, attention mechanisms are performed at the \texttt{Graph Token} level based on the transformed cross codebook, reducing computational complexity while enhancing the model's generalization capability. Extensive experiments on datasets of various sizes demonstrate that VecFormer outperforms the existing Graph Transformer in both performance and speed.
Departures: Distributional Transport for Single-Cell Perturbation Prediction with Neural Schrödinger Bridges
Nov 17, 2025



Predicting single-cell perturbation outcomes directly advances gene function analysis and facilitates drug candidate selection, making it a key driver of both basic and translational biomedical research. However, a major bottleneck in this task is the unpaired nature of single-cell data, as the same cell cannot be observed both before and after perturbation due to the destructive nature of sequencing. Although some neural generative transport models attempt to tackle unpaired single-cell perturbation data, they either lack explicit conditioning or depend on prior spaces for indirect distribution alignment, limiting precise perturbation modeling. In this work, we approximate Schrödinger Bridge (SB), which defines stochastic dynamic mappings recovering the entropy-regularized optimal transport (OT), to directly align the distributions of control and perturbed single-cell populations across different perturbation conditions. Unlike prior SB approximations that rely on bidirectional modeling to infer optimal source-target sample coupling, we leverage Minibatch-OT based pairing to avoid such bidirectional inference and the associated ill-posedness of defining the reverse process. This pairing directly guides bridge learning, yielding a scalable approximation to the SB. We approximate two SB models, one modeling discrete gene activation states and the other continuous expression distributions. Joint training enables accurate perturbation modeling and captures single-cell heterogeneity. Experiments on public genetic and drug perturbation datasets show that our model effectively captures heterogeneous single-cell responses and achieves state-of-the-art performance.
MergeDNA: Context-aware Genome Modeling with Dynamic Tokenization through Token Merging
Nov 17, 2025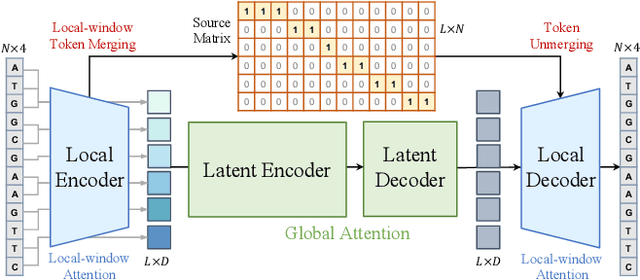
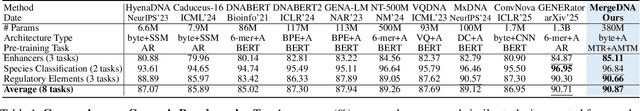
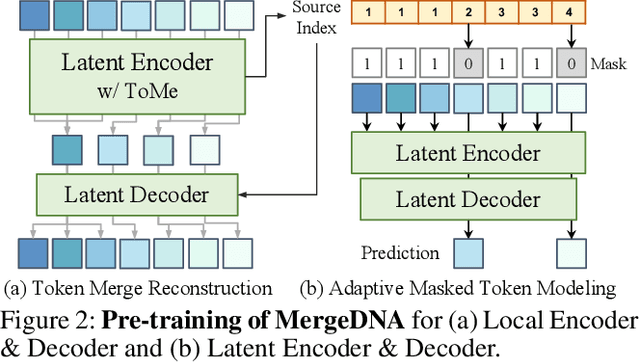
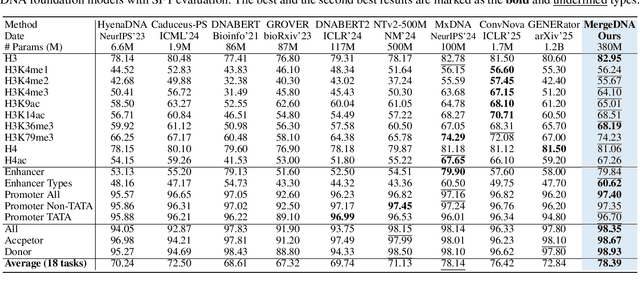
Modeling genomic sequences faces two unsolved challenges: the information density varies widely across different regions, while there is no clearly defined minimum vocabulary unit. Relying on either four primitive bases or independently designed DNA tokenizers, existing approaches with naive masked language modeling pre-training often fail to adapt to the varying complexities of genomic sequences. Leveraging Token Merging techniques, this paper introduces a hierarchical architecture that jointly optimizes a dynamic genomic tokenizer and latent Transformers with context-aware pre-training tasks. As for network structures, the tokenization module automatically chunks adjacent bases into words by stacking multiple layers of the differentiable token merging blocks with local-window constraints, then a Latent Encoder captures the global context of these merged words by full-attention blocks. Symmetrically employing a Latent Decoder and a Local Decoder, MergeDNA learns with two pre-training tasks: Merged Token Reconstruction simultaneously trains the dynamic tokenization module and adaptively filters important tokens, while Adaptive Masked Token Modeling learns to predict these filtered tokens to capture informative contents. Extensive experiments show that MergeDNA achieves superior performance on three popular DNA benchmarks and several multi-omics tasks with fine-tuning or zero-shot evaluation, outperforming typical tokenization methods and large-scale DNA foundation models.
AlphaFold Database Debiasing for Robust Inverse Folding
Jun 10, 2025The AlphaFold Protein Structure Database (AFDB) offers unparalleled structural coverage at near-experimental accuracy, positioning it as a valuable resource for data-driven protein design. However, its direct use in training deep models that are sensitive to fine-grained atomic geometry, such as inverse folding, exposes a critical limitation. Comparative analysis of structural feature distributions reveals that AFDB structures exhibit distinct statistical regularities, reflecting a systematic geometric bias that deviates from the conformational diversity found in experimentally determined structures from the Protein Data Bank (PDB). While AFDB structures are cleaner and more idealized, PDB structures capture the intrinsic variability and physical realism essential for generalization in downstream tasks. To address this discrepancy, we introduce a Debiasing Structure AutoEncoder (DeSAE) that learns to reconstruct native-like conformations from intentionally corrupted backbone geometries. By training the model to recover plausible structural states, DeSAE implicitly captures a more robust and natural structural manifold. At inference, applying DeSAE to AFDB structures produces debiased structures that significantly improve inverse folding performance across multiple benchmarks. This work highlights the critical impact of subtle systematic biases in predicted structures and presents a principled framework for debiasing, significantly boosting the performance of structure-based learning tasks like inverse folding.
Tokenizing Electron Cloud in Protein-Ligand Interaction Learning
May 25, 2025



The affinity and specificity of protein-molecule binding directly impact functional outcomes, uncovering the mechanisms underlying biological regulation and signal transduction. Most deep-learning-based prediction approaches focus on structures of atoms or fragments. However, quantum chemical properties, such as electronic structures, are the key to unveiling interaction patterns but remain largely underexplored. To bridge this gap, we propose ECBind, a method for tokenizing electron cloud signals into quantized embeddings, enabling their integration into downstream tasks such as binding affinity prediction. By incorporating electron densities, ECBind helps uncover binding modes that cannot be fully represented by atom-level models. Specifically, to remove the redundancy inherent in electron cloud signals, a structure-aware transformer and hierarchical codebooks encode 3D binding sites enriched with electron structures into tokens. These tokenized codes are then used for specific tasks with labels. To extend its applicability to a wider range of scenarios, we utilize knowledge distillation to develop an electron-cloud-agnostic prediction model. Experimentally, ECBind demonstrates state-of-the-art performance across multiple tasks, achieving improvements of 6.42\% and 15.58\% in per-structure Pearson and Spearman correlation coefficients, respectively.
GRAPE: Heterogeneous Graph Representation Learning for Genetic Perturbation with Coding and Non-Coding Biotype
May 06, 2025



Predicting genetic perturbations enables the identification of potentially crucial genes prior to wet-lab experiments, significantly improving overall experimental efficiency. Since genes are the foundation of cellular life, building gene regulatory networks (GRN) is essential to understand and predict the effects of genetic perturbations. However, current methods fail to fully leverage gene-related information, and solely rely on simple evaluation metrics to construct coarse-grained GRN. More importantly, they ignore functional differences between biotypes, limiting the ability to capture potential gene interactions. In this work, we leverage pre-trained large language model and DNA sequence model to extract features from gene descriptions and DNA sequence data, respectively, which serve as the initialization for gene representations. Additionally, we introduce gene biotype information for the first time in genetic perturbation, simulating the distinct roles of genes with different biotypes in regulating cellular processes, while capturing implicit gene relationships through graph structure learning (GSL). We propose GRAPE, a heterogeneous graph neural network (HGNN) that leverages gene representations initialized with features from descriptions and sequences, models the distinct roles of genes with different biotypes, and dynamically refines the GRN through GSL. The results on publicly available datasets show that our method achieves state-of-the-art performance.
Adversarial Curriculum Graph-Free Knowledge Distillation for Graph Neural Networks
Apr 02, 2025Data-free Knowledge Distillation (DFKD) is a method that constructs pseudo-samples using a generator without real data, and transfers knowledge from a teacher model to a student by enforcing the student to overcome dimensional differences and learn to mimic the teacher's outputs on these pseudo-samples. In recent years, various studies in the vision domain have made notable advancements in this area. However, the varying topological structures and non-grid nature of graph data render the methods from the vision domain ineffective. Building upon prior research into differentiable methods for graph neural networks, we propose a fast and high-quality data-free knowledge distillation approach in this paper. Without compromising distillation quality, the proposed graph-free KD method (ACGKD) significantly reduces the spatial complexity of pseudo-graphs by leveraging the Binary Concrete distribution to model the graph structure and introducing a spatial complexity tuning parameter. This approach enables efficient gradient computation for the graph structure, thereby accelerating the overall distillation process. Additionally, ACGKD eliminates the dimensional ambiguity between the student and teacher models by increasing the student's dimensions and reusing the teacher's classifier. Moreover, it equips graph knowledge distillation with a CL-based strategy to ensure the student learns graph structures progressively. Extensive experiments demonstrate that ACGKD achieves state-of-the-art performance in distilling knowledge from GNNs without training data.