 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNightjar: Dynamic Adaptive Speculative Decoding for Large Language Models Serving
Dec 27, 2025Speculative decoding (SD) accelerates LLM inference by verifying draft tokens in parallel. However, this method presents a critical trade-off: it improves throughput in low-load, memory-bound systems but degrades performance in high-load, compute-bound environments due to verification overhead. Current SD implementations use a fixed speculative length, failing to adapt to dynamic request rates and creating a significant performance bottleneck in real-world serving scenarios. To overcome this, we propose Nightjar, a novel learning-based algorithm for adaptive speculative inference that adjusts to request load by dynamically selecting the optimal speculative length for different batch sizes and even disabling speculative decoding when it provides no benefit. Experiments show that Nightjar achieves up to 14.8% higher throughput and 20.2% lower latency compared to standard speculative decoding, demonstrating robust efficiency for real-time serving.
A digital SRAM-based compute-in-memory macro for weight-stationary dynamic matrix multiplication in Transformer attention score computation
Nov 15, 2025



Compute-in-memory (CIM) techniques are widely employed in energy-efficient artificial intelligent (AI) processors. They alleviate power and latency bottlenecks caused by extensive data movements between compute and storage units. This work proposes a digital CIM macro to compute Transformer attention. To mitigate dynamic matrix multiplication that is unsuitable for the common weight-stationary CIM paradigm, we reformulate the attention score computation process based on a combined QK-weight matrix, so that inputs can be directly fed to CIM cells to obtain the score results. Moreover, the involved binomial matrix multiplication operation is decomposed into 4 groups of bit-serial shifting and additions, without costly physical multipliers in the CIM. We maximize the energy efficiency of the CIM circuit through zero-value bit-skipping, data-driven word line activation, read-write separate 6T cells and bit-alternating 14T/28T adders. The proposed CIM macro was implemented using a 65-nm process. It occupied only 0.35 mm2 area, and delivered a 42.27 GOPS peak performance with 1.24 mW power consumption at a 1.0 V power supply and a 100 MHz clock frequency, resulting in 34.1 TOPS/W energy efficiency and 120.77 GOPS/mm2 area efficiency. When compared to the CPU and GPU, our CIM macro is 25x and 13x more energy efficient on practical tasks, respectively. Compared with other Transformer-CIMs, our design exhibits at least 7x energy efficiency and at least 2x area efficiency improvements when scaled to the same technology node, showcasing its potential for edge-side intelligent applications.
Can an Easy-to-Hard Curriculum Make Reasoning Emerge in Small Language Models? Evidence from a Four-Stage Curriculum on GPT-2
May 16, 2025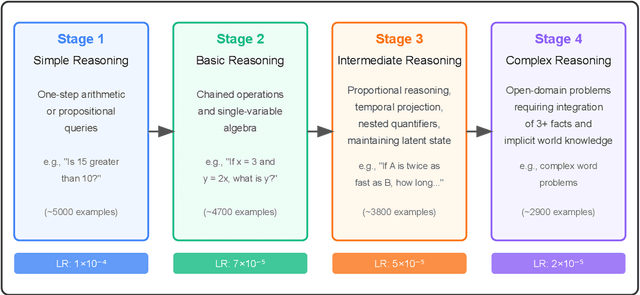



We demonstrate that a developmentally ordered curriculum markedly improves reasoning transparency and sample-efficiency in small language models (SLMs). Concretely, we train Cognivolve, a 124 M-parameter GPT-2 model, on a four-stage syllabus that ascends from lexical matching to multi-step symbolic inference and then evaluate it without any task-specific fine-tuning. Cognivolve reaches target accuracy in half the optimization steps of a single-phase baseline, activates an order-of-magnitude more gradient-salient reasoning heads, and shifts those heads toward deeper layers, yielding higher-entropy attention that balances local and long-range context. The same curriculum applied out of order or with optimizer resets fails to reproduce these gains, confirming that progression--not extra compute--drives the effect. We also identify open challenges: final-answer success still lags a conventional run by about 30%, and our saliency probe under-detects verbal-knowledge heads in the hardest stage, suggesting directions for mixed-stage fine-tuning and probe expansion.
Adjoint Sampling: Highly Scalable Diffusion Samplers via Adjoint Matching
Apr 16, 2025We introduce Adjoint Sampling, a highly scalable and efficient algorithm for learning diffusion processes that sample from unnormalized densities, or energy functions. It is the first on-policy approach that allows significantly more gradient updates than the number of energy evaluations and model samples, allowing us to scale to much larger problem settings than previously explored by similar methods. Our framework is theoretically grounded in stochastic optimal control and shares the same theoretical guarantees as Adjoint Matching, being able to train without the need for corrective measures that push samples towards the target distribution. We show how to incorporate key symmetries, as well as periodic boundary conditions, for modeling molecules in both cartesian and torsional coordinates. We demonstrate the effectiveness of our approach through extensive experiments on classical energy functions, and further scale up to neural network-based energy models where we perform amortized conformer generation across many molecular systems. To encourage further research in developing highly scalable sampling methods, we plan to open source these challenging benchmarks, where successful methods can directly impact progress in computational chemistry.
M2IV: Towards Efficient and Fine-grained Multimodal In-Context Learning in Large Vision-Language Models
Apr 06, 2025Multimodal in-context learning (ICL) is a vital capability for Large Vision-Language Models (LVLMs), allowing task adaptation via contextual prompts without parameter retraining. However, its application is hindered by the token-intensive nature of inputs and the high complexity of cross-modal few-shot learning, which limits the expressive power of representation methods. To tackle these challenges, we propose \textbf{M2IV}, a method that substitutes explicit demonstrations with learnable \textbf{I}n-context \textbf{V}ectors directly integrated into LVLMs. By exploiting the complementary strengths of multi-head attention (\textbf{M}HA) and multi-layer perceptrons (\textbf{M}LP), M2IV achieves robust cross-modal fidelity and fine-grained semantic distillation through training. This significantly enhances performance across diverse LVLMs and tasks and scales efficiently to many-shot scenarios, bypassing the context window limitations. We also introduce \textbf{VLibrary}, a repository for storing and retrieving M2IV, enabling flexible LVLM steering for tasks like cross-modal alignment, customized generation and safety improvement. Experiments across seven benchmarks and three LVLMs show that M2IV surpasses Vanilla ICL and prior representation engineering approaches, with an average accuracy gain of \textbf{3.74\%} over ICL with the same shot count, alongside substantial efficiency advantages.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025



The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
All-atom Diffusion Transformers: Unified generative modelling of molecules and materials
Mar 05, 2025



Diffusion models are the standard toolkit for generative modelling of 3D atomic systems. However, for different types of atomic systems - such as molecules and materials - the generative processes are usually highly specific to the target system despite the underlying physics being the same. We introduce the All-atom Diffusion Transformer (ADiT), a unified latent diffusion framework for jointly generating both periodic materials and non-periodic molecular systems using the same model: (1) An autoencoder maps a unified, all-atom representations of molecules and materials to a shared latent embedding space; and (2) A diffusion model is trained to generate new latent embeddings that the autoencoder can decode to sample new molecules or materials. Experiments on QM9 and MP20 datasets demonstrate that jointly trained ADiT generates realistic and valid molecules as well as materials, exceeding state-of-the-art results from molecule and crystal-specific models. ADiT uses standard Transformers for both the autoencoder and diffusion model, resulting in significant speedups during training and inference compared to equivariant diffusion models. Scaling ADiT up to half a billion parameters predictably improves performance, representing a step towards broadly generalizable foundation models for generative chemistry. Open source code: https://github.com/facebookresearch/all-atom-diffusion-transformer
Learning Smooth and Expressive Interatomic Potentials for Physical Property Prediction
Feb 17, 2025



Machine learning interatomic potentials (MLIPs) have become increasingly effective at approximating quantum mechanical calculations at a fraction of the computational cost. However, lower errors on held out test sets do not always translate to improved results on downstream physical property prediction tasks. In this paper, we propose testing MLIPs on their practical ability to conserve energy during molecular dynamic simulations. If passed, improved correlations are found between test errors and their performance on physical property prediction tasks. We identify choices which may lead to models failing this test, and use these observations to improve upon highly-expressive models. The resulting model, eSEN, provides state-of-the-art results on a range of physical property prediction tasks, including materials stability prediction, thermal conductivity prediction, and phonon calculations.
CeViT: Copula-Enhanced Vision Transformer in multi-task learning and bi-group image covariates with an application to myopia screening
Jan 11, 2025



We aim to assist image-based myopia screening by resolving two longstanding problems, "how to integrate the information of ocular images of a pair of eyes" and "how to incorporate the inherent dependence among high-myopia status and axial length for both eyes." The classification-regression task is modeled as a novel 4-dimensional muti-response regression, where discrete responses are allowed, that relates to two dependent 3rd-order tensors (3D ultrawide-field fundus images). We present a Vision Transformer-based bi-channel architecture, named CeViT, where the common features of a pair of eyes are extracted via a shared Transformer encoder, and the interocular asymmetries are modeled through separated multilayer perceptron heads. Statistically, we model the conditional dependence among mixture of discrete-continuous responses given the image covariates by a so-called copula loss. We establish a new theoretical framework regarding fine-tuning on CeViT based on latent representations, allowing the black-box fine-tuning procedure interpretable and guaranteeing higher relative efficiency of fine-tuning weight estimation in the asymptotic setting. We apply CeViT to an annotated ultrawide-field fundus image dataset collected by Shanghai Eye \& ENT Hospital, demonstrating that CeViT enhances the baseline model in both accuracy of classifying high-myopia and prediction of AL on both eyes.
Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
Oct 16, 2024



The ability to discover new materials with desirable properties is critical for numerous applications from helping mitigate climate change to advances in next generation computing hardware. AI has the potential to accelerate materials discovery and design by more effectively exploring the chemical space compared to other computational methods or by trial-and-error. While substantial progress has been made on AI for materials data, benchmarks, and models, a barrier that has emerged is the lack of publicly available training data and open pre-trained models. To address this, we present a Meta FAIR release of the Open Materials 2024 (OMat24) large-scale open dataset and an accompanying set of pre-trained models. OMat24 contains over 110 million density functional theory (DFT) calculations focused on structural and compositional diversity. Our EquiformerV2 models achieve state-of-the-art performance on the Matbench Discovery leaderboard and are capable of predicting ground-state stability and formation energies to an F1 score above 0.9 and an accuracy of 20 meV/atom, respectively. We explore the impact of model size, auxiliary denoising objectives, and fine-tuning on performance across a range of datasets including OMat24, MPtraj, and Alexandria. The open release of the OMat24 dataset and models enables the research community to build upon our efforts and drive further advancements in AI-assisted materials science.