 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Smooth and Expressive Interatomic Potentials for Physical Property Prediction
Feb 17, 2025



Machine learning interatomic potentials (MLIPs) have become increasingly effective at approximating quantum mechanical calculations at a fraction of the computational cost. However, lower errors on held out test sets do not always translate to improved results on downstream physical property prediction tasks. In this paper, we propose testing MLIPs on their practical ability to conserve energy during molecular dynamic simulations. If passed, improved correlations are found between test errors and their performance on physical property prediction tasks. We identify choices which may lead to models failing this test, and use these observations to improve upon highly-expressive models. The resulting model, eSEN, provides state-of-the-art results on a range of physical property prediction tasks, including materials stability prediction, thermal conductivity prediction, and phonon calculations.
Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
Oct 16, 2024



The ability to discover new materials with desirable properties is critical for numerous applications from helping mitigate climate change to advances in next generation computing hardware. AI has the potential to accelerate materials discovery and design by more effectively exploring the chemical space compared to other computational methods or by trial-and-error. While substantial progress has been made on AI for materials data, benchmarks, and models, a barrier that has emerged is the lack of publicly available training data and open pre-trained models. To address this, we present a Meta FAIR release of the Open Materials 2024 (OMat24) large-scale open dataset and an accompanying set of pre-trained models. OMat24 contains over 110 million density functional theory (DFT) calculations focused on structural and compositional diversity. Our EquiformerV2 models achieve state-of-the-art performance on the Matbench Discovery leaderboard and are capable of predicting ground-state stability and formation energies to an F1 score above 0.9 and an accuracy of 20 meV/atom, respectively. We explore the impact of model size, auxiliary denoising objectives, and fine-tuning on performance across a range of datasets including OMat24, MPtraj, and Alexandria. The open release of the OMat24 dataset and models enables the research community to build upon our efforts and drive further advancements in AI-assisted materials science.
Fine-Tuned Language Models Generate Stable Inorganic Materials as Text
Feb 06, 2024We propose fine-tuning large language models for generation of stable materials. While unorthodox, fine-tuning large language models on text-encoded atomistic data is simple to implement yet reliable, with around 90% of sampled structures obeying physical constraints on atom positions and charges. Using energy above hull calculations from both learned ML potentials and gold-standard DFT calculations, we show that our strongest model (fine-tuned LLaMA-2 70B) can generate materials predicted to be metastable at about twice the rate (49% vs 28%) of CDVAE, a competing diffusion model. Because of text prompting's inherent flexibility, our models can simultaneously be used for unconditional generation of stable material, infilling of partial structures and text-conditional generation. Finally, we show that language models' ability to capture key symmetries of crystal structures improves with model scale, suggesting that the biases of pretrained LLMs are surprisingly well-suited for atomistic data.
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Oct 25, 2023Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of $\sim$120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks.
Reducing SO Convolutions to SO for Efficient Equivariant GNNs
Feb 07, 2023Graph neural networks that model 3D data, such as point clouds or atoms, are typically desired to be $SO(3)$ equivariant, i.e., equivariant to 3D rotations. Unfortunately equivariant convolutions, which are a fundamental operation for equivariant networks, increase significantly in computational complexity as higher-order tensors are used. In this paper, we address this issue by reducing the $SO(3)$ convolutions or tensor products to mathematically equivalent convolutions in $SO(2)$ . This is accomplished by aligning the node embeddings' primary axis with the edge vectors, which sparsifies the tensor product and reduces the computational complexity from $O(L^6)$ to $O(L^3)$, where $L$ is the degree of the representation. We demonstrate the potential implications of this improvement by proposing the Equivariant Spherical Channel Network (eSCN), a graph neural network utilizing our novel approach to equivariant convolutions, which achieves state-of-the-art results on the large-scale OC-20 dataset.
AdsorbML: Accelerating Adsorption Energy Calculations with Machine Learning
Nov 29, 2022Computational catalysis is playing an increasingly significant role in the design of catalysts across a wide range of applications. A common task for many computational methods is the need to accurately compute the minimum binding energy - the adsorption energy - for an adsorbate and a catalyst surface of interest. Traditionally, the identification of low energy adsorbate-surface configurations relies on heuristic methods and researcher intuition. As the desire to perform high-throughput screening increases, it becomes challenging to use heuristics and intuition alone. In this paper, we demonstrate machine learning potentials can be leveraged to identify low energy adsorbate-surface configurations more accurately and efficiently. Our algorithm provides a spectrum of trade-offs between accuracy and efficiency, with one balanced option finding the lowest energy configuration, within a 0.1 eV threshold, 86.63% of the time, while achieving a 1387x speedup in computation. To standardize benchmarking, we introduce the Open Catalyst Dense dataset containing nearly 1,000 diverse surfaces and 87,045 unique configurations.
Spherical Channels for Modeling Atomic Interactions
Jun 29, 2022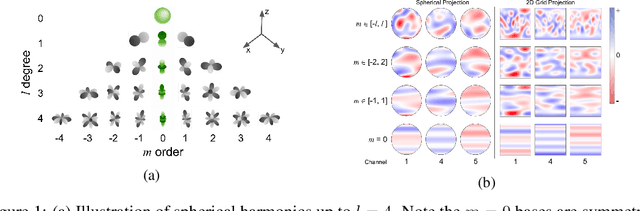
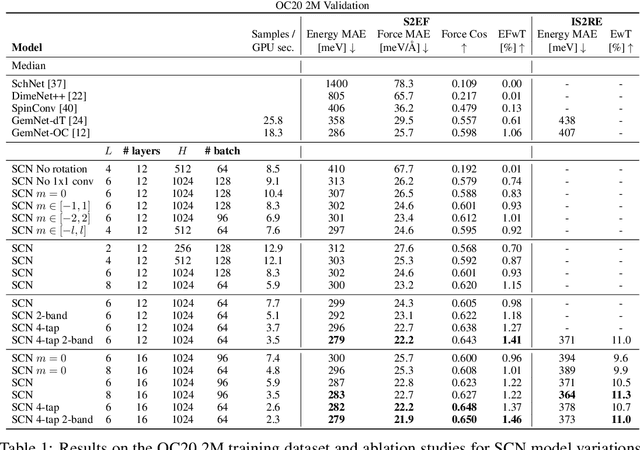

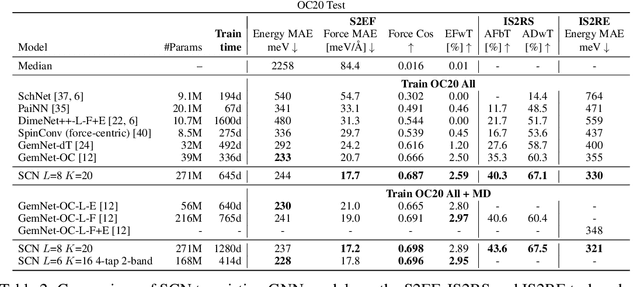
Modeling the energy and forces of atomic systems is a fundamental problem in computational chemistry with the potential to help address many of the world's most pressing problems, including those related to energy scarcity and climate change. These calculations are traditionally performed using Density Functional Theory, which is computationally very expensive. Machine learning has the potential to dramatically improve the efficiency of these calculations from days or hours to seconds. We propose the Spherical Channel Network (SCN) to model atomic energies and forces. The SCN is a graph neural network where nodes represent atoms and edges their neighboring atoms. The atom embeddings are a set of spherical functions, called spherical channels, represented using spherical harmonics. We demonstrate, that by rotating the embeddings based on the 3D edge orientation, more information may be utilized while maintaining the rotational equivariance of the messages. While equivariance is a desirable property, we find that by relaxing this constraint in both message passing and aggregation, improved accuracy may be achieved. We demonstrate state-of-the-art results on the large-scale Open Catalyst 2020 dataset in both energy and force prediction for numerous tasks and metrics.
The Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Jun 17, 2022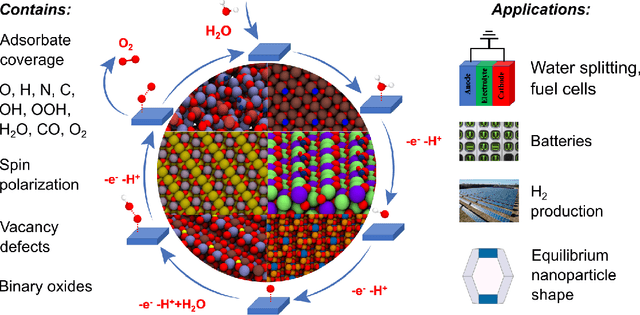
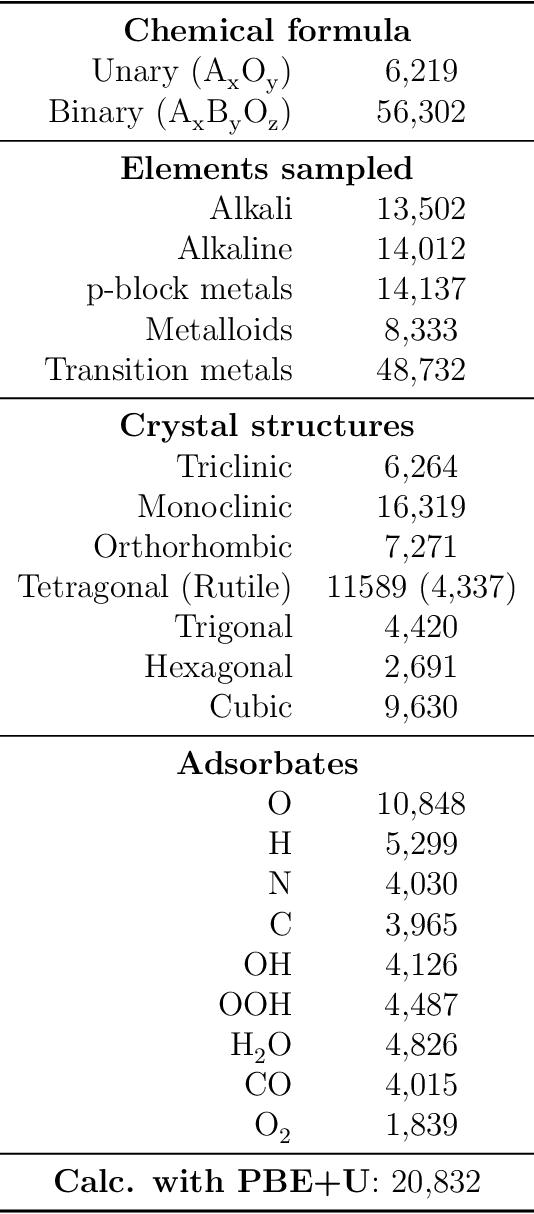
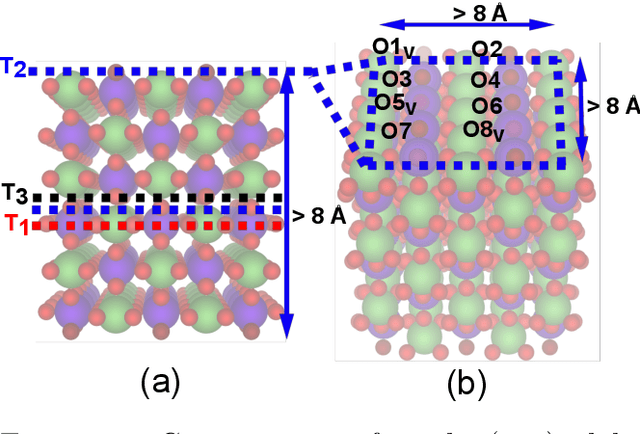
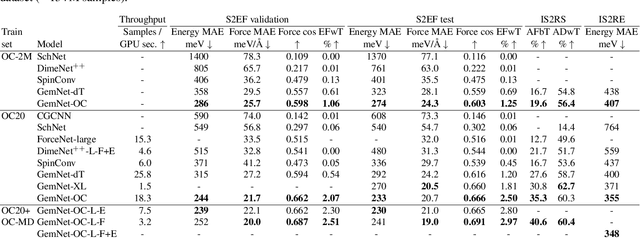
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.
How Do Graph Networks Generalize to Large and Diverse Molecular Systems?
Apr 06, 2022
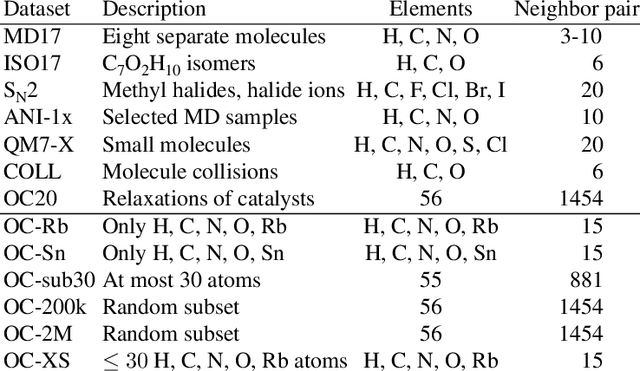
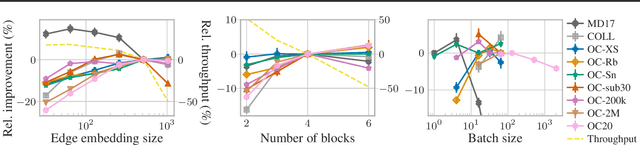
The predominant method of demonstrating progress of atomic graph neural networks are benchmarks on small and limited datasets. The implicit hypothesis behind this approach is that progress on these narrow datasets generalize to the large diversity of chemistry. This generalizability would be very helpful for research, but currently remains untested. In this work we test this assumption by identifying four aspects of complexity in which many datasets are lacking: 1. Chemical diversity (number of different elements), 2. system size (number of atoms per sample), 3. dataset size (number of data samples), and 4. domain shift (similarity of the training and test set). We introduce multiple subsets of the large Open Catalyst 2020 (OC20) dataset to independently investigate each of these aspects. We then perform 21 ablation studies and sensitivity analyses on 9 datasets testing both previously proposed and new model enhancements. We find that some improvements are consistent between datasets, but many are not and some even have opposite effects. Based on this analysis, we identify a smaller dataset that correlates well with the full OC20 dataset, and propose the GemNet-OC model, which outperforms the previous state-of-the-art on OC20 by 16%, while reducing training time by a factor of 10. Overall, our findings challenge the common belief that graph neural networks work equally well independent of dataset size and diversity, and suggest that caution must be exercised when making generalizations based on narrow datasets.
Towards Training Billion Parameter Graph Neural Networks for Atomic Simulations
Mar 18, 2022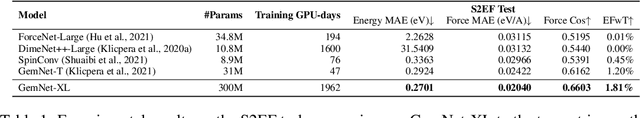
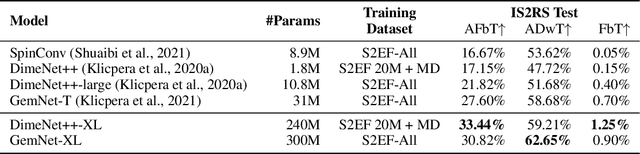
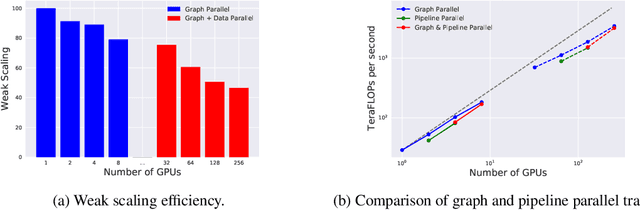
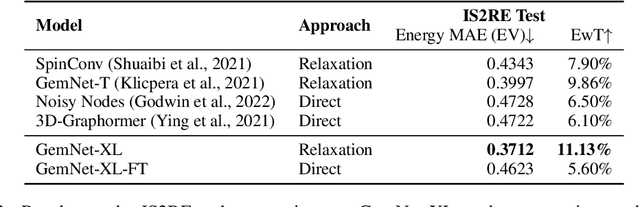
Recent progress in Graph Neural Networks (GNNs) for modeling atomic simulations has the potential to revolutionize catalyst discovery, which is a key step in making progress towards the energy breakthroughs needed to combat climate change. However, the GNNs that have proven most effective for this task are memory intensive as they model higher-order interactions in the graphs such as those between triplets or quadruplets of atoms, making it challenging to scale these models. In this paper, we introduce Graph Parallelism, a method to distribute input graphs across multiple GPUs, enabling us to train very large GNNs with hundreds of millions or billions of parameters. We empirically evaluate our method by scaling up the number of parameters of the recently proposed DimeNet++ and GemNet models by over an order of magnitude. On the large-scale Open Catalyst 2020 (OC20) dataset, these graph-parallelized models lead to relative improvements of 1) 15% on the force MAE metric for the S2EF task and 2) 21% on the AFbT metric for the IS2RS task, establishing new state-of-the-art results.