 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEmpowering Polymeric Materials Discovery by Artificial Intelligence
Jun 18, 2026Polymeric materials underpin modern technologies spanning energy storage, microelectronics, healthcare and sustainable manufacturing. Yet their rational design remains exceptionally challenging because material performance emerges from complex interactions among molecular composition, chain architecture, processing history and hierarchical structural evolution across multiple length and time scales. Consequently, polymer research has long relied on labor-intensive experimentation and fragmented modeling approaches, limiting both mechanistic understanding and innovation efficiency. Recent advances in data infrastructure, machine learning, large artificial intelligence (AI) models and laboratory automation are beginning to reshape this landscape. Rather than functioning as isolated tools, polymer databases, predictive models, AI agents and automated laboratories are increasingly converging into interconnected discovery ecosystems. As a result, the central challenge is shifting from improving predictive accuracy alone to enabling reliable decision-making, adaptive learning and seamless integration across computation, experimentation and scientific reasoning. We argue that polymer science is entering an era of autonomous discovery, in which data, simulation, reasoning and experimentation operate within self-improving feedback loops that continuously generate hypotheses, design materials, execute experiments and refine predictive models. By unifying molecular design, process optimization, experimental validation and industrial translation, such autonomous ecosystems establish a more predictive, reproducible and scalable paradigm for polymer innovation, fundamentally transforming how polymer research is conducted.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025



The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Leveraging CORAL-Correlation Consistency Network for Semi-Supervised Left Atrium MRI Segmentation
Oct 21, 2024



Semi-supervised learning (SSL) has been widely used to learn from both a few labeled images and many unlabeled images to overcome the scarcity of labeled samples in medical image segmentation. Most current SSL-based segmentation methods use pixel values directly to identify similar features in labeled and unlabeled data. They usually fail to accurately capture the intricate attachment structures in the left atrium, such as the areas of inconsistent density or exhibit outward curvatures, adding to the complexity of the task. In this paper, we delve into this issue and introduce an effective solution, CORAL(Correlation-Aligned)-Correlation Consistency Network (CORN), to capture the global structure shape and local details of Left Atrium. Diverging from previous methods focused on each local pixel value, the CORAL-Correlation Consistency Module (CCM) in the CORN leverages second-order statistical information to capture global structural features by minimizing the distribution discrepancy between labeled and unlabeled samples in feature space. Yet, direct construction of features from unlabeled data frequently results in ``Sample Selection Bias'', leading to flawed supervision. We thus further propose the Dynamic Feature Pool (DFP) for the CCM, which utilizes a confidence-based filtering strategy to remove incorrectly selected features and regularize both teacher and student models by constraining the similarity matrix to be consistent. Extensive experiments on the Left Atrium dataset have shown that the proposed CORN outperforms previous state-of-the-art semi-supervised learning methods.
M$^3$-Impute: Mask-guided Representation Learning for Missing Value Imputation
Oct 11, 2024



Missing values are a common problem that poses significant challenges to data analysis and machine learning. This problem necessitates the development of an effective imputation method to fill in the missing values accurately, thereby enhancing the overall quality and utility of the datasets. Existing imputation methods, however, fall short of explicitly considering the `missingness' information in the data during the embedding initialization stage and modeling the entangled feature and sample correlations during the learning process, thus leading to inferior performance. We propose M$^3$-Impute, which aims to explicitly leverage the missingness information and such correlations with novel masking schemes. M$^3$-Impute first models the data as a bipartite graph and uses a graph neural network to learn node embeddings, where the refined embedding initialization process directly incorporates the missingness information. They are then optimized through M$^3$-Impute's novel feature correlation unit (FRU) and sample correlation unit (SRU) that effectively captures feature and sample correlations for imputation. Experiment results on 25 benchmark datasets under three different missingness settings show the effectiveness of M$^3$-Impute by achieving 20 best and 4 second-best MAE scores on average.
DAE-Fuse: An Adaptive Discriminative Autoencoder for Multi-Modality Image Fusion
Sep 16, 2024



Multi-modality image fusion aims to integrate complementary data information from different imaging modalities into a single image. Existing methods often generate either blurry fused images that lose fine-grained semantic information or unnatural fused images that appear perceptually cropped from the inputs. In this work, we propose a novel two-phase discriminative autoencoder framework, termed DAE-Fuse, that generates sharp and natural fused images. In the adversarial feature extraction phase, we introduce two discriminative blocks into the encoder-decoder architecture, providing an additional adversarial loss to better guide feature extraction by reconstructing the source images. While the two discriminative blocks are adapted in the attention-guided cross-modality fusion phase to distinguish the structural differences between the fused output and the source inputs, injecting more naturalness into the results. Extensive experiments on public infrared-visible, medical image fusion, and downstream object detection datasets demonstrate our method's superiority and generalizability in both quantitative and qualitative evaluations.
Forces are not Enough: Benchmark and Critical Evaluation for Machine Learning Force Fields with Molecular Simulations
Oct 13, 2022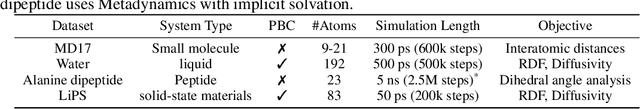
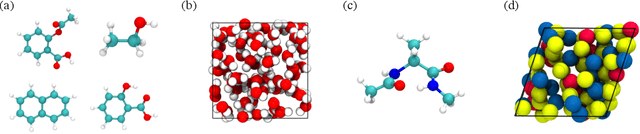

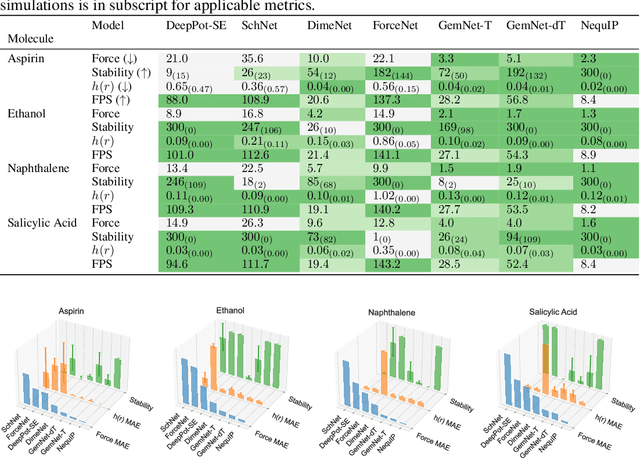
Molecular dynamics (MD) simulation techniques are widely used for various natural science applications. Increasingly, machine learning (ML) force field (FF) models begin to replace ab-initio simulations by predicting forces directly from atomic structures. Despite significant progress in this area, such techniques are primarily benchmarked by their force/energy prediction errors, even though the practical use case would be to produce realistic MD trajectories. We aim to fill this gap by introducing a novel benchmark suite for ML MD simulation. We curate representative MD systems, including water, organic molecules, peptide, and materials, and design evaluation metrics corresponding to the scientific objectives of respective systems. We benchmark a collection of state-of-the-art (SOTA) ML FF models and illustrate, in particular, how the commonly benchmarked force accuracy is not well aligned with relevant simulation metrics. We demonstrate when and how selected SOTA methods fail, along with offering directions for further improvement. Specifically, we identify stability as a key metric for ML models to improve. Our benchmark suite comes with a comprehensive open-source codebase for training and simulation with ML FFs to facilitate further work.
Learning Pair Potentials using Differentiable Simulations
Sep 16, 2022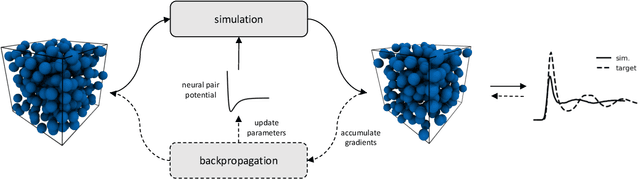
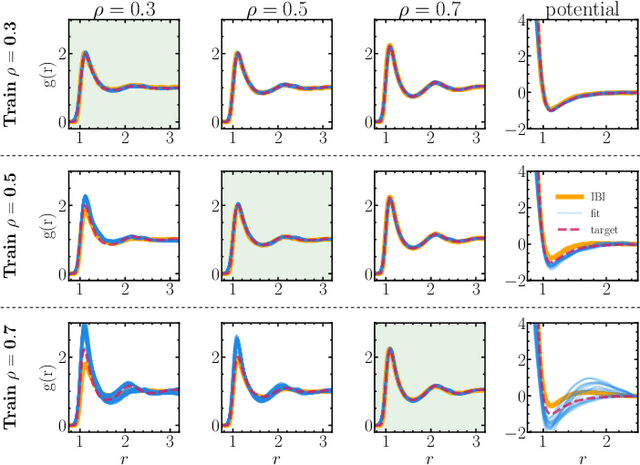
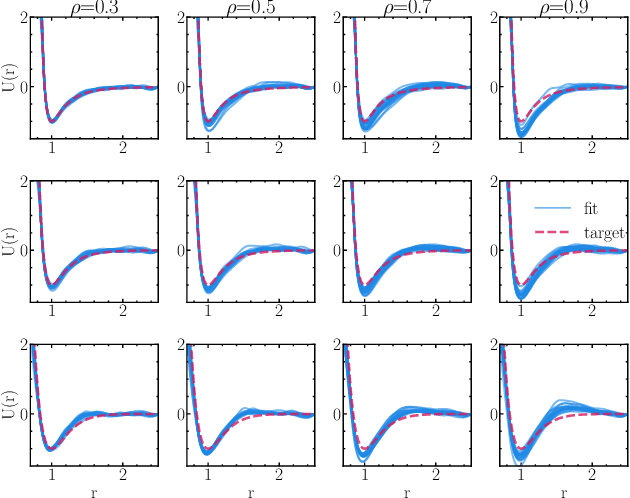
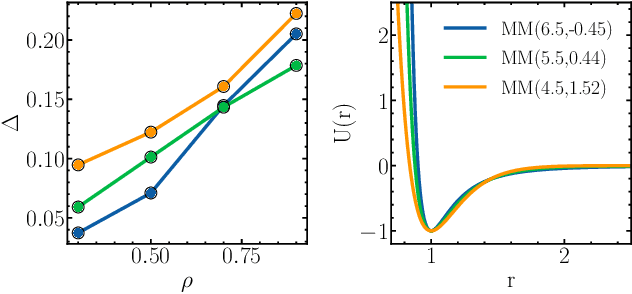
Learning pair interactions from experimental or simulation data is of great interest for molecular simulations. We propose a general stochastic method for learning pair interactions from data using differentiable simulations (DiffSim). DiffSim defines a loss function based on structural observables, such as the radial distribution function, through molecular dynamics (MD) simulations. The interaction potentials are then learned directly by stochastic gradient descent, using backpropagation to calculate the gradient of the structural loss metric with respect to the interaction potential through the MD simulation. This gradient-based method is flexible and can be configured to simulate and optimize multiple systems simultaneously. For example, it is possible to simultaneously learn potentials for different temperatures or for different compositions. We demonstrate the approach by recovering simple pair potentials, such as Lennard-Jones systems, from radial distribution functions. We find that DiffSim can be used to probe a wider functional space of pair potentials compared to traditional methods like Iterative Boltzmann Inversion. We show that our methods can be used to simultaneously fit potentials for simulations at different compositions and temperatures to improve the transferability of the learned potentials.
LenAtten: An Effective Length Controlling Unit For Text Summarization
Jun 01, 2021
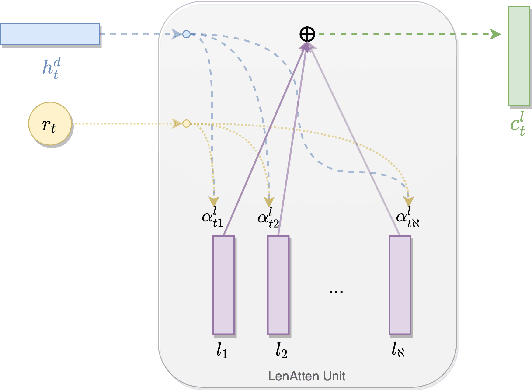
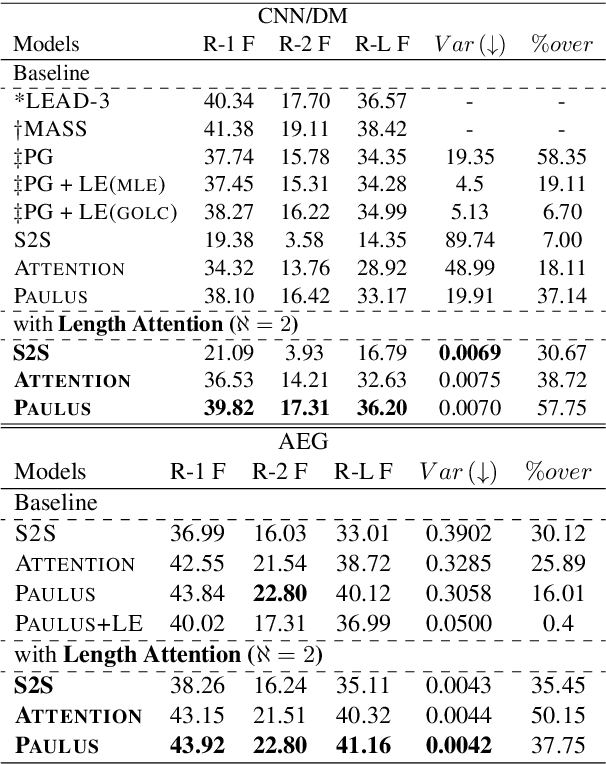
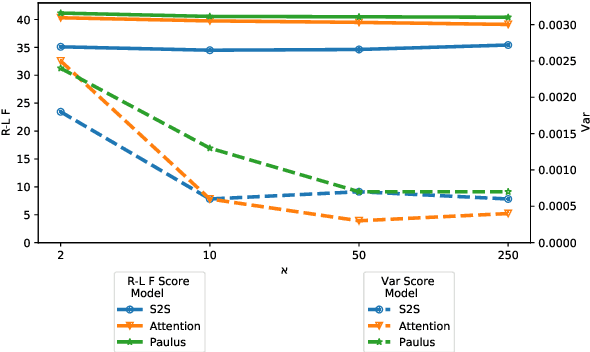
Fixed length summarization aims at generating summaries with a preset number of words or characters. Most recent researches incorporate length information with word embeddings as the input to the recurrent decoding unit, causing a compromise between length controllability and summary quality. In this work, we present an effective length controlling unit Length Attention (LenAtten) to break this trade-off. Experimental results show that LenAtten not only brings improvements in length controllability and ROGUE scores but also has great generalization ability. In the task of generating a summary with the target length, our model is 732 times better than the best-performing length controllable summarizer in length controllability on the CNN/Daily Mail dataset.