 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdjoint Sampling: Highly Scalable Diffusion Samplers via Adjoint Matching
Apr 16, 2025We introduce Adjoint Sampling, a highly scalable and efficient algorithm for learning diffusion processes that sample from unnormalized densities, or energy functions. It is the first on-policy approach that allows significantly more gradient updates than the number of energy evaluations and model samples, allowing us to scale to much larger problem settings than previously explored by similar methods. Our framework is theoretically grounded in stochastic optimal control and shares the same theoretical guarantees as Adjoint Matching, being able to train without the need for corrective measures that push samples towards the target distribution. We show how to incorporate key symmetries, as well as periodic boundary conditions, for modeling molecules in both cartesian and torsional coordinates. We demonstrate the effectiveness of our approach through extensive experiments on classical energy functions, and further scale up to neural network-based energy models where we perform amortized conformer generation across many molecular systems. To encourage further research in developing highly scalable sampling methods, we plan to open source these challenging benchmarks, where successful methods can directly impact progress in computational chemistry.
All-atom Diffusion Transformers: Unified generative modelling of molecules and materials
Mar 05, 2025



Diffusion models are the standard toolkit for generative modelling of 3D atomic systems. However, for different types of atomic systems - such as molecules and materials - the generative processes are usually highly specific to the target system despite the underlying physics being the same. We introduce the All-atom Diffusion Transformer (ADiT), a unified latent diffusion framework for jointly generating both periodic materials and non-periodic molecular systems using the same model: (1) An autoencoder maps a unified, all-atom representations of molecules and materials to a shared latent embedding space; and (2) A diffusion model is trained to generate new latent embeddings that the autoencoder can decode to sample new molecules or materials. Experiments on QM9 and MP20 datasets demonstrate that jointly trained ADiT generates realistic and valid molecules as well as materials, exceeding state-of-the-art results from molecule and crystal-specific models. ADiT uses standard Transformers for both the autoencoder and diffusion model, resulting in significant speedups during training and inference compared to equivariant diffusion models. Scaling ADiT up to half a billion parameters predictably improves performance, representing a step towards broadly generalizable foundation models for generative chemistry. Open source code: https://github.com/facebookresearch/all-atom-diffusion-transformer
Flow Matching with General Discrete Paths: A Kinetic-Optimal Perspective
Dec 04, 2024The design space of discrete-space diffusion or flow generative models are significantly less well-understood than their continuous-space counterparts, with many works focusing only on a simple masked construction. In this work, we aim to take a holistic approach to the construction of discrete generative models based on continuous-time Markov chains, and for the first time, allow the use of arbitrary discrete probability paths, or colloquially, corruption processes. Through the lens of optimizing the symmetric kinetic energy, we propose velocity formulas that can be applied to any given probability path, completely decoupling the probability and velocity, and giving the user the freedom to specify any desirable probability path based on expert knowledge specific to the data domain. Furthermore, we find that a special construction of mixture probability paths optimizes the symmetric kinetic energy for the discrete case. We empirically validate the usefulness of this new design space across multiple modalities: text generation, inorganic material generation, and image generation. We find that we can outperform the mask construction even in text with kinetic-optimal mixture paths, while we can make use of domain-specific constructions of the probability path over the visual domain.
FlowLLM: Flow Matching for Material Generation with Large Language Models as Base Distributions
Oct 30, 2024Material discovery is a critical area of research with the potential to revolutionize various fields, including carbon capture, renewable energy, and electronics. However, the immense scale of the chemical space makes it challenging to explore all possible materials experimentally. In this paper, we introduce FlowLLM, a novel generative model that combines large language models (LLMs) and Riemannian flow matching (RFM) to design novel crystalline materials. FlowLLM first fine-tunes an LLM to learn an effective base distribution of meta-stable crystals in a text representation. After converting to a graph representation, the RFM model takes samples from the LLM and iteratively refines the coordinates and lattice parameters. Our approach significantly outperforms state-of-the-art methods, increasing the generation rate of stable materials by over three times and increasing the rate for stable, unique, and novel crystals by $\sim50\%$ - a huge improvement on a difficult problem. Additionally, the crystals generated by FlowLLM are much closer to their relaxed state when compared with another leading model, significantly reducing post-hoc computational cost.
Distribution Learning for Molecular Regression
Jul 30, 2024



Using "soft" targets to improve model performance has been shown to be effective in classification settings, but the usage of soft targets for regression is a much less studied topic in machine learning. The existing literature on the usage of soft targets for regression fails to properly assess the method's limitations, and empirical evaluation is quite limited. In this work, we assess the strengths and drawbacks of existing methods when applied to molecular property regression tasks. Our assessment outlines key biases present in existing methods and proposes methods to address them, evaluated through careful ablation studies. We leverage these insights to propose Distributional Mixture of Experts (DMoE): A model-independent, and data-independent method for regression which trains a model to predict probability distributions of its targets. Our proposed loss function combines the cross entropy between predicted and target distributions and the L1 distance between their expected values to produce a loss function that is robust to the outlined biases. We evaluate the performance of DMoE on different molecular property prediction datasets -- Open Catalyst (OC20), MD17, and QM9 -- across different backbone model architectures -- SchNet, GemNet, and Graphormer. Our results demonstrate that the proposed method is a promising alternative to classical regression for molecular property prediction tasks, showing improvements over baselines on all datasets and architectures.
FlowMM: Generating Materials with Riemannian Flow Matching
Jun 07, 2024Crystalline materials are a fundamental component in next-generation technologies, yet modeling their distribution presents unique computational challenges. Of the plausible arrangements of atoms in a periodic lattice only a vanishingly small percentage are thermodynamically stable, which is a key indicator of the materials that can be experimentally realized. Two fundamental tasks in this area are to (a) predict the stable crystal structure of a known composition of elements and (b) propose novel compositions along with their stable structures. We present FlowMM, a pair of generative models that achieve state-of-the-art performance on both tasks while being more efficient and more flexible than competing methods. We generalize Riemannian Flow Matching to suit the symmetries inherent to crystals: translation, rotation, permutation, and periodic boundary conditions. Our framework enables the freedom to choose the flow base distributions, drastically simplifying the problem of learning crystal structures compared with diffusion models. In addition to standard benchmarks, we validate FlowMM's generated structures with quantum chemistry calculations, demonstrating that it is about 3x more efficient, in terms of integration steps, at finding stable materials compared to previous open methods.
* https://github.com/facebookresearch/flowmm
Fine-Tuned Language Models Generate Stable Inorganic Materials as Text
Feb 06, 2024We propose fine-tuning large language models for generation of stable materials. While unorthodox, fine-tuning large language models on text-encoded atomistic data is simple to implement yet reliable, with around 90% of sampled structures obeying physical constraints on atom positions and charges. Using energy above hull calculations from both learned ML potentials and gold-standard DFT calculations, we show that our strongest model (fine-tuned LLaMA-2 70B) can generate materials predicted to be metastable at about twice the rate (49% vs 28%) of CDVAE, a competing diffusion model. Because of text prompting's inherent flexibility, our models can simultaneously be used for unconditional generation of stable material, infilling of partial structures and text-conditional generation. Finally, we show that language models' ability to capture key symmetries of crystal structures improves with model scale, suggesting that the biases of pretrained LLMs are surprisingly well-suited for atomistic data.
The Open DAC 2023 Dataset and Challenges for Sorbent Discovery in Direct Air Capture
Nov 01, 2023



New methods for carbon dioxide removal are urgently needed to combat global climate change. Direct air capture (DAC) is an emerging technology to capture carbon dioxide directly from ambient air. Metal-organic frameworks (MOFs) have been widely studied as potentially customizable adsorbents for DAC. However, discovering promising MOF sorbents for DAC is challenging because of the vast chemical space to explore and the need to understand materials as functions of humidity and temperature. We explore a computational approach benefiting from recent innovations in machine learning (ML) and present a dataset named Open DAC 2023 (ODAC23) consisting of more than 38M density functional theory (DFT) calculations on more than 8,800 MOF materials containing adsorbed CO2 and/or H2O. ODAC23 is by far the largest dataset of MOF adsorption calculations at the DFT level of accuracy currently available. In addition to probing properties of adsorbed molecules, the dataset is a rich source of information on structural relaxation of MOFs, which will be useful in many contexts beyond specific applications for DAC. A large number of MOFs with promising properties for DAC are identified directly in ODAC23. We also trained state-of-the-art ML models on this dataset to approximate calculations at the DFT level. This open-source dataset and our initial ML models will provide an important baseline for future efforts to identify MOFs for a wide range of applications, including DAC.
Spherical Channels for Modeling Atomic Interactions
Jun 29, 2022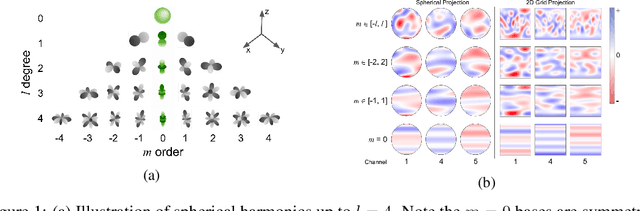
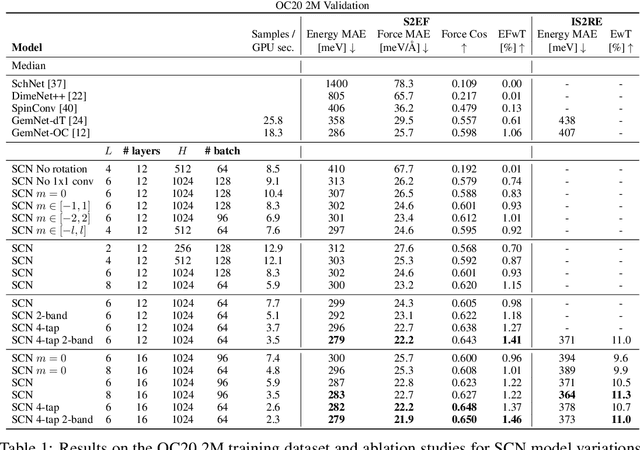

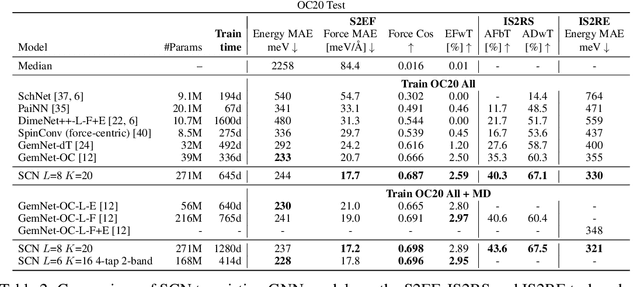
Modeling the energy and forces of atomic systems is a fundamental problem in computational chemistry with the potential to help address many of the world's most pressing problems, including those related to energy scarcity and climate change. These calculations are traditionally performed using Density Functional Theory, which is computationally very expensive. Machine learning has the potential to dramatically improve the efficiency of these calculations from days or hours to seconds. We propose the Spherical Channel Network (SCN) to model atomic energies and forces. The SCN is a graph neural network where nodes represent atoms and edges their neighboring atoms. The atom embeddings are a set of spherical functions, called spherical channels, represented using spherical harmonics. We demonstrate, that by rotating the embeddings based on the 3D edge orientation, more information may be utilized while maintaining the rotational equivariance of the messages. While equivariance is a desirable property, we find that by relaxing this constraint in both message passing and aggregation, improved accuracy may be achieved. We demonstrate state-of-the-art results on the large-scale Open Catalyst 2020 dataset in both energy and force prediction for numerous tasks and metrics.
Wav2Vec-Aug: Improved self-supervised training with limited data
Jun 27, 2022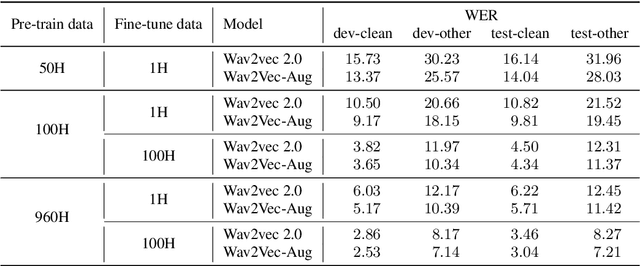
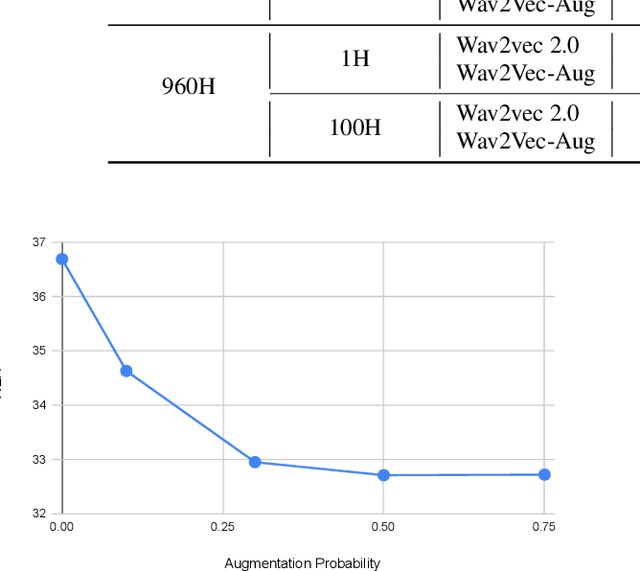
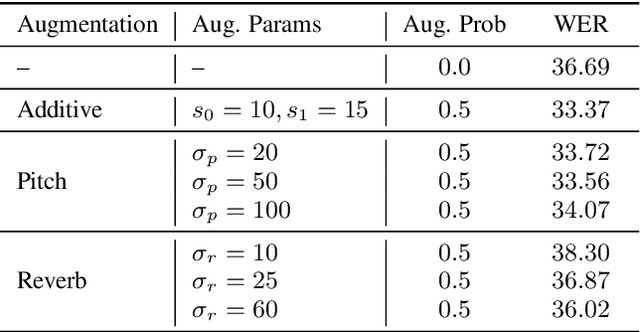
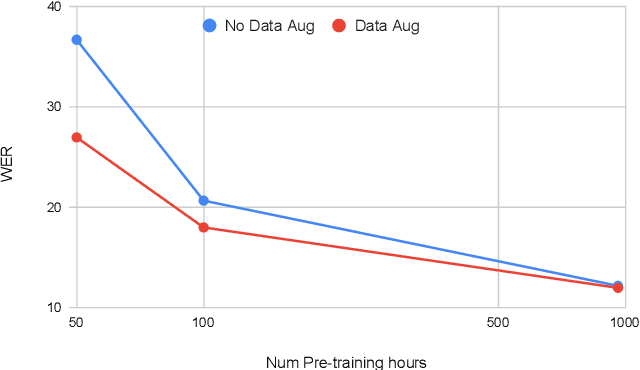
Self-supervised learning (SSL) of speech representations has received much attention over the last few years but most work has focused on languages and domains with an abundance of unlabeled data. However, for many languages there is a shortage even in the unlabeled data which limits the effectiveness of SSL. In this work, we focus on the problem of applying SSL to domains with limited available data by leveraging data augmentation for Wav2Vec 2.0 pretraining. Further, we propose improvements to each component of the model which result in a combined relative word error rate (WER) improvement of up to 13% compared to Wav2Vec 2.0 on Librispeech test-clean / other.