 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobotic Grasping from Classical to Modern: A Survey
Feb 08, 2022



Robotic Grasping has always been an active topic in robotics since grasping is one of the fundamental but most challenging skills of robots. It demands the coordination of robotic perception, planning, and control for robustness and intelligence. However, current solutions are still far behind humans, especially when confronting unstructured scenarios. In this paper, we survey the advances of robotic grasping, starting from the classical formulations and solutions to the modern ones. By reviewing the history of robotic grasping, we want to provide a complete view of this community, and perhaps inspire the combination and fusion of different ideas, which we think would be helpful to touch and explore the essence of robotic grasping problems. In detail, we firstly give an overview of the analytic methods for robotic grasping. After that, we provide a discussion on the recent state-of-the-art data-driven grasping approaches rising in recent years. With the development of computer vision, semantic grasping is being widely investigated and can be the basis of intelligent manipulation and skill learning for autonomous robotic systems in the future. Therefore, in our survey, we also briefly review the recent progress in this topic. Finally, we discuss the open problems and the future research directions that may be important for the human-level robustness, autonomy, and intelligence of robots.
Generative Coarse-Graining of Molecular Conformations
Jan 28, 2022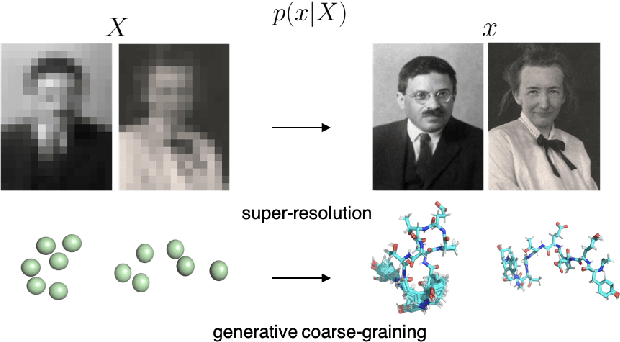

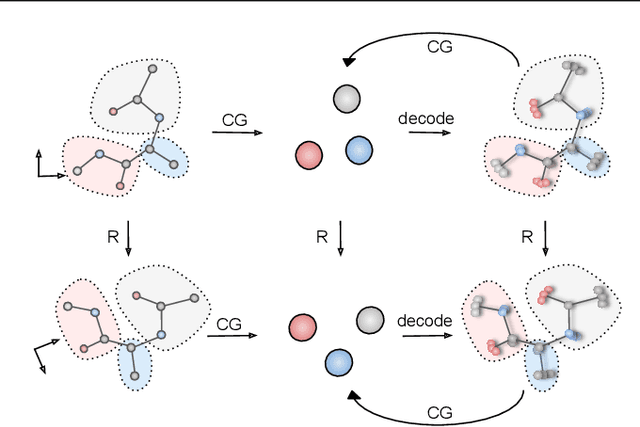
Coarse-graining (CG) of molecular simulations simplifies the particle representation by grouping selected atoms into pseudo-beads and therefore drastically accelerates simulation. However, such CG procedure induces information losses, which makes accurate backmapping, i.e., restoring fine-grained (FG) coordinates from CG coordinates, a long-standing challenge. Inspired by the recent progress in generative models and equivariant networks, we propose a novel model that rigorously embeds the vital probabilistic nature and geometric consistency requirements of the backmapping transformation. Our model encodes the FG uncertainties into an invariant latent space and decodes them back to FG geometries via equivariant convolutions. To standardize the evaluation of this domain, we further provide three comprehensive benchmarks based on molecular dynamics trajectories. Extensive experiments show that our approach always recovers more realistic structures and outperforms existing data-driven methods with a significant margin.
LegoDNN: Block-grained Scaling of Deep Neural Networks for Mobile Vision
Dec 18, 2021
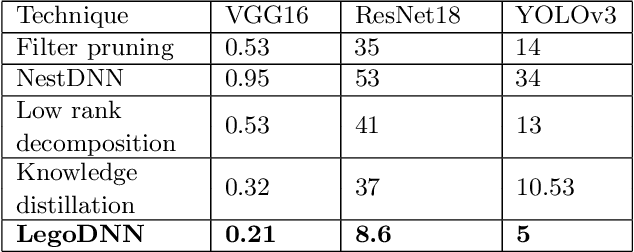
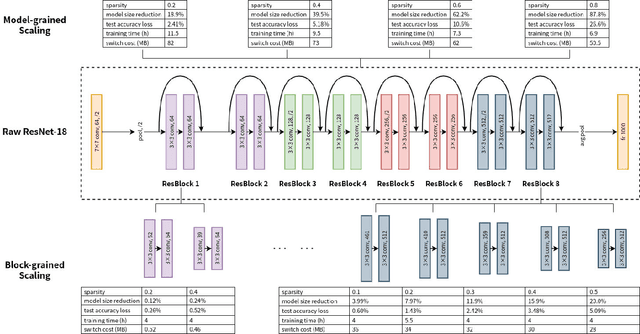
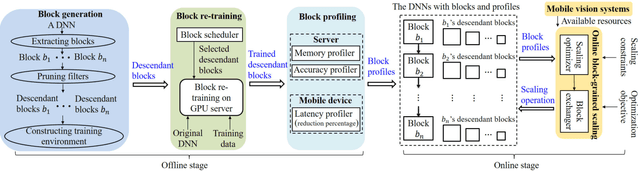
Deep neural networks (DNNs) have become ubiquitous techniques in mobile and embedded systems for applications such as image/object recognition and classification. The trend of executing multiple DNNs simultaneously exacerbate the existing limitations of meeting stringent latency/accuracy requirements on resource constrained mobile devices. The prior art sheds light on exploring the accuracy-resource tradeoff by scaling the model sizes in accordance to resource dynamics. However, such model scaling approaches face to imminent challenges: (i) large space exploration of model sizes, and (ii) prohibitively long training time for different model combinations. In this paper, we present LegoDNN, a lightweight, block-grained scaling solution for running multi-DNN workloads in mobile vision systems. LegoDNN guarantees short model training times by only extracting and training a small number of common blocks (e.g. 5 in VGG and 8 in ResNet) in a DNN. At run-time, LegoDNN optimally combines the descendant models of these blocks to maximize accuracy under specific resources and latency constraints, while reducing switching overhead via smart block-level scaling of the DNN. We implement LegoDNN in TensorFlow Lite and extensively evaluate it against state-of-the-art techniques (FLOP scaling, knowledge distillation and model compression) using a set of 12 popular DNN models. Evaluation results show that LegoDNN provides 1,296x to 279,936x more options in model sizes without increasing training time, thus achieving as much as 31.74% improvement in inference accuracy and 71.07% reduction in scaling energy consumptions.
* 13 pages, 15 figures
Implications of Topological Imbalance for Representation Learning on Biomedical Knowledge Graphs
Dec 13, 2021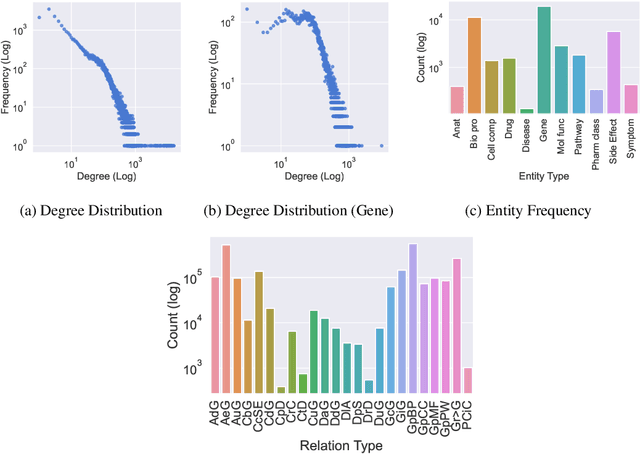
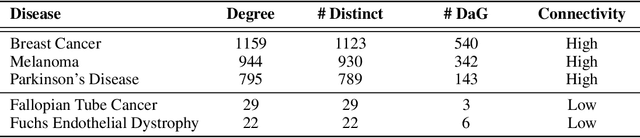
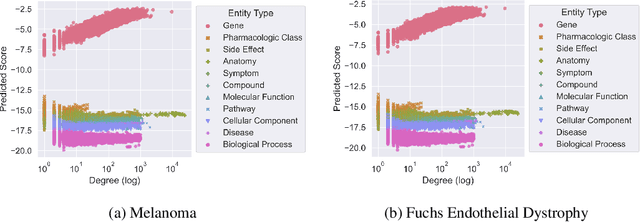
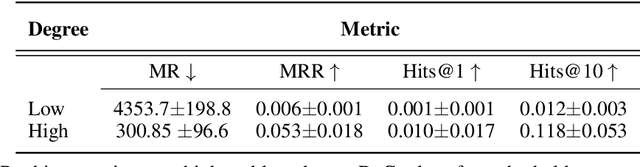
Improving on the standard of care for diseases is predicated on better treatments, which in turn relies on finding and developing new drugs. However, drug discovery is a complex and costly process. Adoption of methods from machine learning has given rise to creation of drug discovery knowledge graphs which utilize the inherent interconnected nature of the domain. Graph-based data modelling, combined with knowledge graph embeddings provide a more intuitive representation of the domain and are suitable for inference tasks such as predicting missing links. One such example would be producing ranked lists of likely associated genes for a given disease, often referred to as target discovery. It is thus critical that these predictions are not only pertinent but also biologically meaningful. However, knowledge graphs can be biased either directly due to the underlying data sources that are integrated or due to modeling choices in the construction of the graph, one consequence of which is that certain entities can get topologically overrepresented. We show how knowledge graph embedding models can be affected by this structural imbalance, resulting in densely connected entities being highly ranked no matter the context. We provide support for this observation across different datasets, models and predictive tasks. Further, we show how the graph topology can be perturbed to artificially alter the rank of a gene via random, biologically meaningless information. This suggests that such models can be more influenced by the frequency of entities rather than biological information encoded in the relations, creating issues when entity frequency is not a true reflection of underlying data. Our results highlight the importance of data modeling choices and emphasizes the need for practitioners to be mindful of these issues when interpreting model outputs and during knowledge graph composition.
Make A Long Image Short: Adaptive Token Length for Vision Transformers
Dec 06, 2021The vision transformer splits each image into a sequence of tokens with fixed length and processes the tokens in the same way as words in natural language processing. More tokens normally lead to better performance but considerably increased computational cost. Motivated by the proverb "A picture is worth a thousand words" we aim to accelerate the ViT model by making a long image short. To this end, we propose a novel approach to assign token length adaptively during inference. Specifically, we first train a ViT model, called Resizable-ViT (ReViT), that can process any given input with diverse token lengths. Then, we retrieve the "token-length label" from ReViT and use it to train a lightweight Token-Length Assigner (TLA). The token-length labels are the smallest number of tokens to split an image that the ReViT can make the correct prediction, and TLA is learned to allocate the optimal token length based on these labels. The TLA enables the ReViT to process the image with the minimum sufficient number of tokens during inference. Thus, the inference speed is boosted by reducing the token numbers in the ViT model. Our approach is general and compatible with modern vision transformer architectures and can significantly reduce computational expanse. We verified the effectiveness of our methods on multiple representative ViT models (DeiT, LV-ViT, and TimesFormer) across two tasks (image classification and action recognition).
Training BatchNorm Only in Neural Architecture Search and Beyond
Dec 01, 2021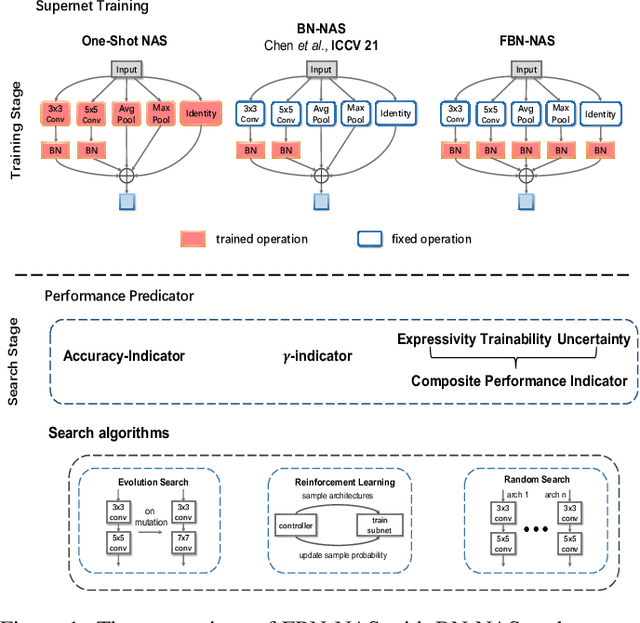

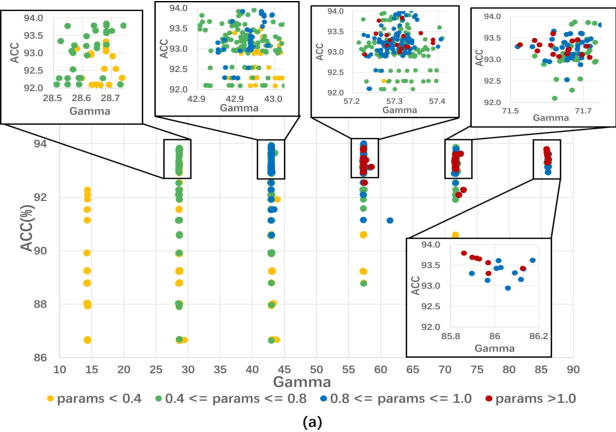
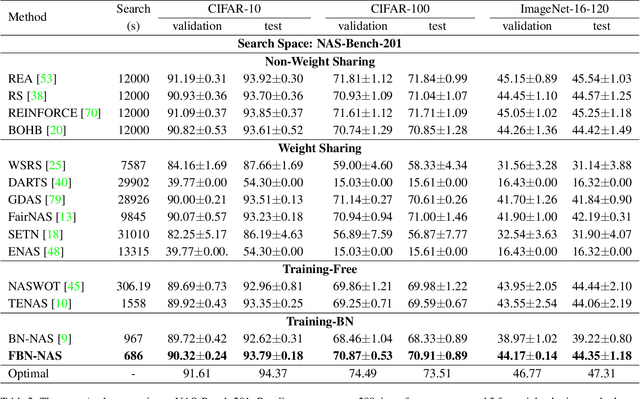
This work investigates the usage of batch normalization in neural architecture search (NAS). Specifically, Frankle et al. find that training BatchNorm only can achieve nontrivial performance. Furthermore, Chen et al. claim that training BatchNorm only can speed up the training of the one-shot NAS supernet over ten times. Critically, there is no effort to understand 1) why training BatchNorm only can find the perform-well architectures with the reduced supernet-training time, and 2) what is the difference between the train-BN-only supernet and the standard-train supernet. We begin by showing that the train-BN-only networks converge to the neural tangent kernel regime, obtain the same training dynamics as train all parameters theoretically. Our proof supports the claim to train BatchNorm only on supernet with less training time. Then, we empirically disclose that train-BN-only supernet provides an advantage on convolutions over other operators, cause unfair competition between architectures. This is due to only the convolution operator being attached with BatchNorm. Through experiments, we show that such unfairness makes the search algorithm prone to select models with convolutions. To solve this issue, we introduce fairness in the search space by placing a BatchNorm layer on every operator. However, we observe that the performance predictor in Chen et al. is inapplicable on the new search space. To this end, we propose a novel composite performance indicator to evaluate networks from three perspectives: expressivity, trainability, and uncertainty, derived from the theoretical property of BatchNorm. We demonstrate the effectiveness of our approach on multiple NAS-benchmarks (NAS-Bench101, NAS-Bench-201) and search spaces (DARTS search space and MobileNet search space).
How to transfer algorithmic reasoning knowledge to learn new algorithms?
Oct 26, 2021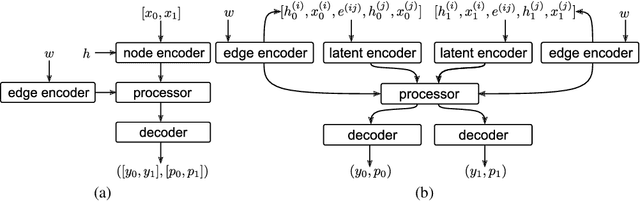
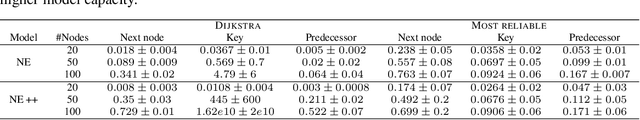
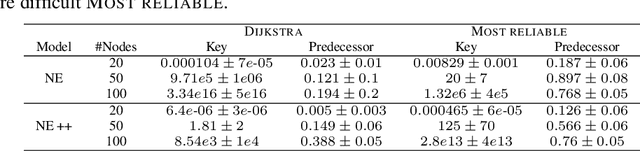
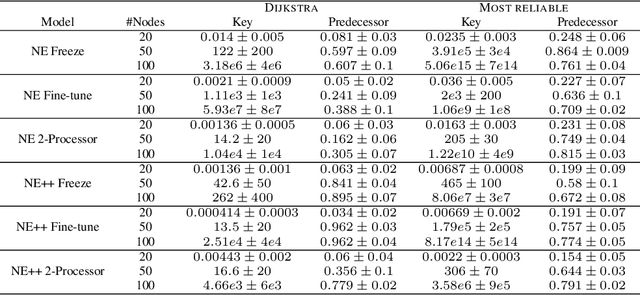
Learning to execute algorithms is a fundamental problem that has been widely studied. Prior work~\cite{veli19neural} has shown that to enable systematic generalisation on graph algorithms it is critical to have access to the intermediate steps of the program/algorithm. In many reasoning tasks, where algorithmic-style reasoning is important, we only have access to the input and output examples. Thus, inspired by the success of pre-training on similar tasks or data in Natural Language Processing (NLP) and Computer Vision, we set out to study how we can transfer algorithmic reasoning knowledge. Specifically, we investigate how we can use algorithms for which we have access to the execution trace to learn to solve similar tasks for which we do not. We investigate two major classes of graph algorithms, parallel algorithms such as breadth-first search and Bellman-Ford and sequential greedy algorithms such as Prim and Dijkstra. Due to the fundamental differences between algorithmic reasoning knowledge and feature extractors such as used in Computer Vision or NLP, we hypothesise that standard transfer techniques will not be sufficient to achieve systematic generalisation. To investigate this empirically we create a dataset including 9 algorithms and 3 different graph types. We validate this empirically and show how instead multi-task learning can be used to achieve the transfer of algorithmic reasoning knowledge.
CATRO: Channel Pruning via Class-Aware Trace Ratio Optimization
Oct 21, 2021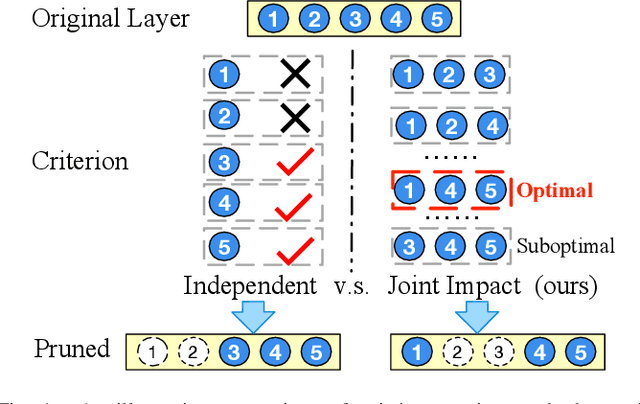
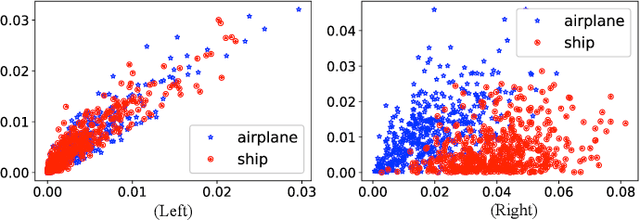
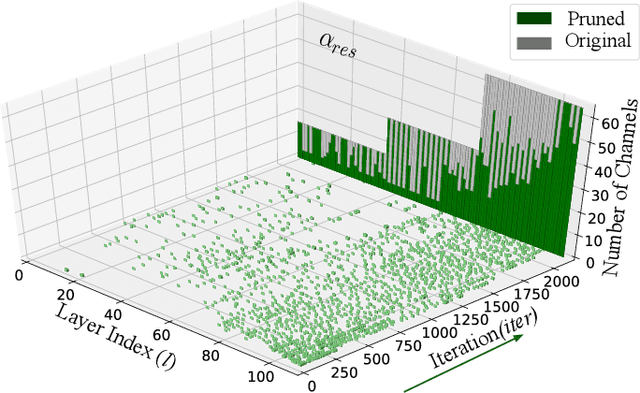
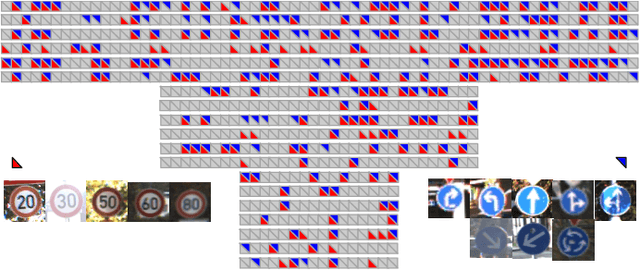
Deep convolutional neural networks are shown to be overkill with high parametric and computational redundancy in many application scenarios, and an increasing number of works have explored model pruning to obtain lightweight and efficient networks. However, most existing pruning approaches are driven by empirical heuristics and rarely consider the joint impact of channels, leading to unguaranteed and suboptimal performance. In this paper, we propose a novel channel pruning method via class-aware trace ratio optimization (CATRO) to reduce the computational burden and accelerate the model inference. Utilizing class information from a few samples, CATRO measures the joint impact of multiple channels by feature space discriminations and consolidates the layer-wise impact of preserved channels. By formulating channel pruning as a submodular set function maximization problem, CATRO solves it efficiently via a two-stage greedy iterative optimization procedure. More importantly, we present theoretical justifications on convergence and performance of CATRO. Experimental results demonstrate that CATRO achieves higher accuracy with similar computation cost or lower computation cost with similar accuracy than other state-of-the-art channel pruning algorithms. In addition, because of its class-aware property, CATRO is suitable to prune efficient networks adaptively for various classification subtasks, enhancing handy deployment and usage of deep networks in real-world applications.
Neural Algorithmic Reasoners are Implicit Planners
Oct 11, 2021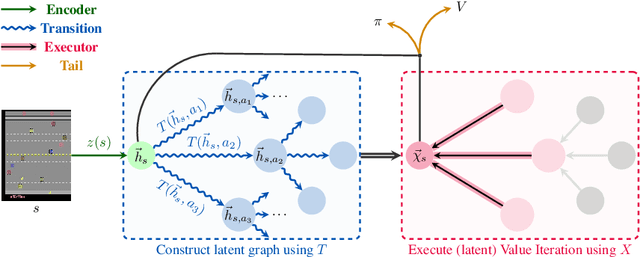
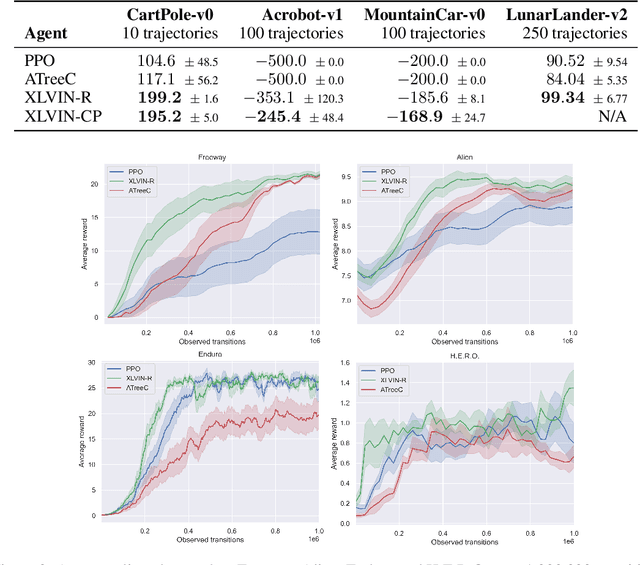

Implicit planning has emerged as an elegant technique for combining learned models of the world with end-to-end model-free reinforcement learning. We study the class of implicit planners inspired by value iteration, an algorithm that is guaranteed to yield perfect policies in fully-specified tabular environments. We find that prior approaches either assume that the environment is provided in such a tabular form -- which is highly restrictive -- or infer "local neighbourhoods" of states to run value iteration over -- for which we discover an algorithmic bottleneck effect. This effect is caused by explicitly running the planning algorithm based on scalar predictions in every state, which can be harmful to data efficiency if such scalars are improperly predicted. We propose eXecuted Latent Value Iteration Networks (XLVINs), which alleviate the above limitations. Our method performs all planning computations in a high-dimensional latent space, breaking the algorithmic bottleneck. It maintains alignment with value iteration by carefully leveraging neural graph-algorithmic reasoning and contrastive self-supervised learning. Across eight low-data settings -- including classical control, navigation and Atari -- XLVINs provide significant improvements to data efficiency against value iteration-based implicit planners, as well as relevant model-free baselines. Lastly, we empirically verify that XLVINs can closely align with value iteration.
Pre-training Molecular Graph Representation with 3D Geometry
Oct 07, 2021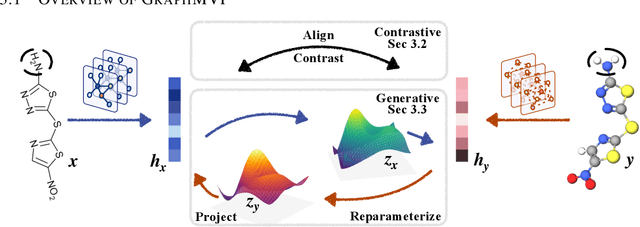
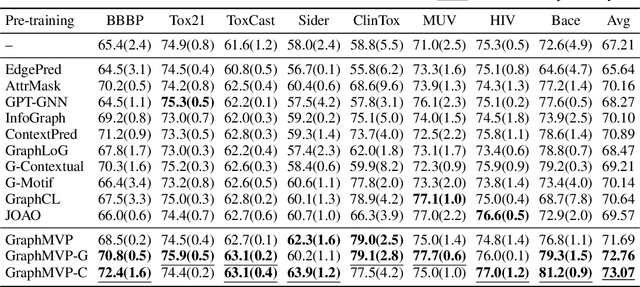

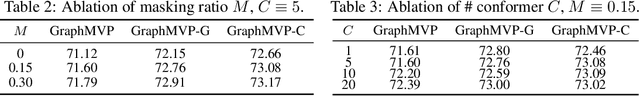
Molecular graph representation learning is a fundamental problem in modern drug and material discovery. Molecular graphs are typically modeled by their 2D topological structures, but it has been recently discovered that 3D geometric information plays a more vital role in predicting molecular functionalities. However, the lack of 3D information in real-world scenarios has significantly impeded the learning of geometric graph representation. To cope with this challenge, we propose the Graph Multi-View Pre-training (GraphMVP) framework where self-supervised learning (SSL) is performed by leveraging the correspondence and consistency between 2D topological structures and 3D geometric views. GraphMVP effectively learns a 2D molecular graph encoder that is enhanced by richer and more discriminative 3D geometry. We further provide theoretical insights to justify the effectiveness of GraphMVP. Finally, comprehensive experiments show that GraphMVP can consistently outperform existing graph SSL methods.