 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Role of Graph Topology in the Performance of Biomedical Knowledge Graph Completion Models
Sep 06, 2024



Knowledge Graph Completion has been increasingly adopted as a useful method for several tasks in biomedical research, like drug repurposing or drug-target identification. To that end, a variety of datasets and Knowledge Graph Embedding models has been proposed over the years. However, little is known about the properties that render a dataset useful for a given task and, even though theoretical properties of Knowledge Graph Embedding models are well understood, their practical utility in this field remains controversial. We conduct a comprehensive investigation into the topological properties of publicly available biomedical Knowledge Graphs and establish links to the accuracy observed in real-world applications. By releasing all model predictions and a new suite of analysis tools we invite the community to build upon our work and continue improving the understanding of these crucial applications.
Implications of Topological Imbalance for Representation Learning on Biomedical Knowledge Graphs
Dec 13, 2021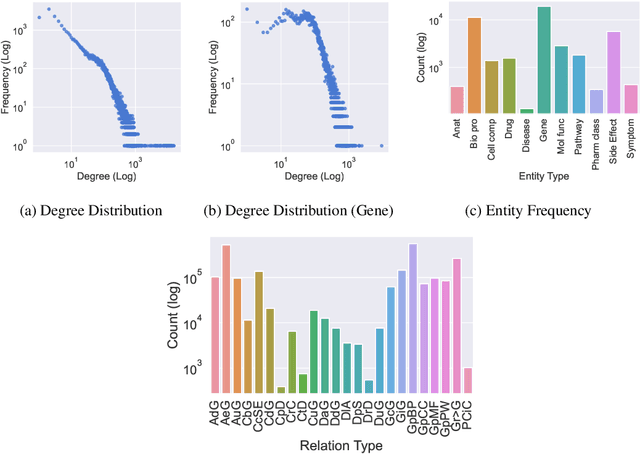
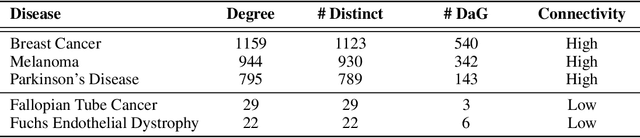
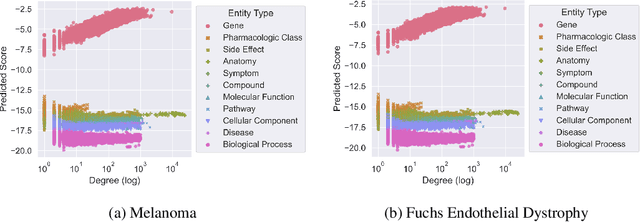
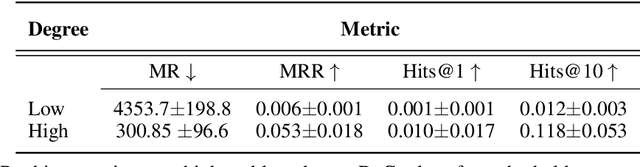
Improving on the standard of care for diseases is predicated on better treatments, which in turn relies on finding and developing new drugs. However, drug discovery is a complex and costly process. Adoption of methods from machine learning has given rise to creation of drug discovery knowledge graphs which utilize the inherent interconnected nature of the domain. Graph-based data modelling, combined with knowledge graph embeddings provide a more intuitive representation of the domain and are suitable for inference tasks such as predicting missing links. One such example would be producing ranked lists of likely associated genes for a given disease, often referred to as target discovery. It is thus critical that these predictions are not only pertinent but also biologically meaningful. However, knowledge graphs can be biased either directly due to the underlying data sources that are integrated or due to modeling choices in the construction of the graph, one consequence of which is that certain entities can get topologically overrepresented. We show how knowledge graph embedding models can be affected by this structural imbalance, resulting in densely connected entities being highly ranked no matter the context. We provide support for this observation across different datasets, models and predictive tasks. Further, we show how the graph topology can be perturbed to artificially alter the rank of a gene via random, biologically meaningless information. This suggests that such models can be more influenced by the frequency of entities rather than biological information encoded in the relations, creating issues when entity frequency is not a true reflection of underlying data. Our results highlight the importance of data modeling choices and emphasizes the need for practitioners to be mindful of these issues when interpreting model outputs and during knowledge graph composition.
A Unified View of Relational Deep Learning for Drug Pair Scoring
Nov 14, 2021
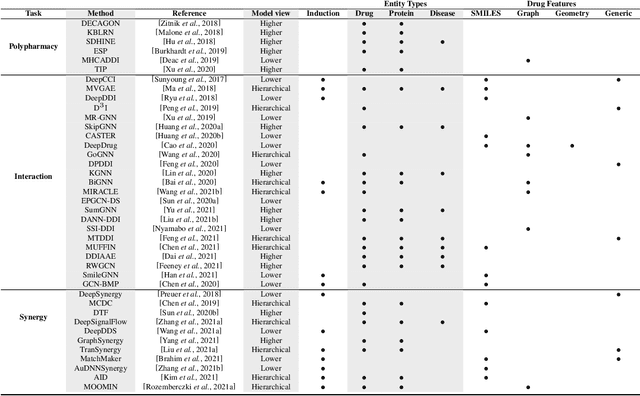
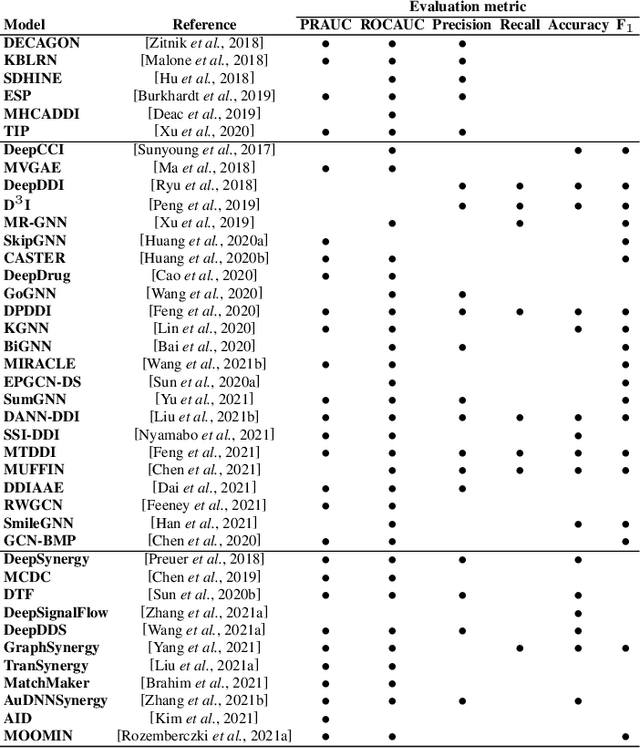
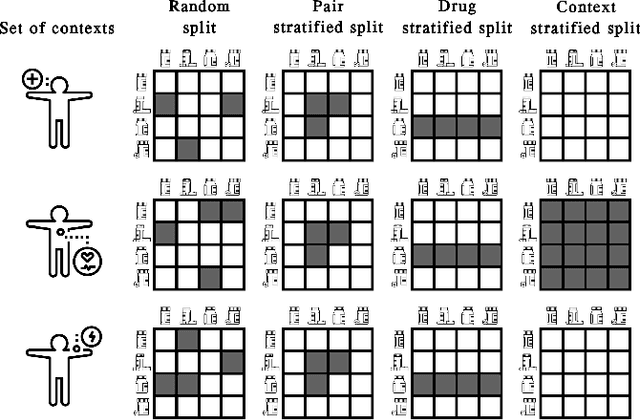
In recent years, numerous machine learning models which attempt to solve polypharmacy side effect identification, drug-drug interaction prediction and combination therapy design tasks have been proposed. Here, we present a unified theoretical view of relational machine learning models which can address these tasks. We provide fundamental definitions, compare existing model architectures and discuss performance metrics, datasets and evaluation protocols. In addition, we emphasize possible high impact applications and important future research directions in this domain.
Understanding the Performance of Knowledge Graph Embeddings in Drug Discovery
Jun 07, 2021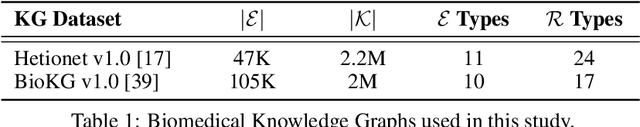
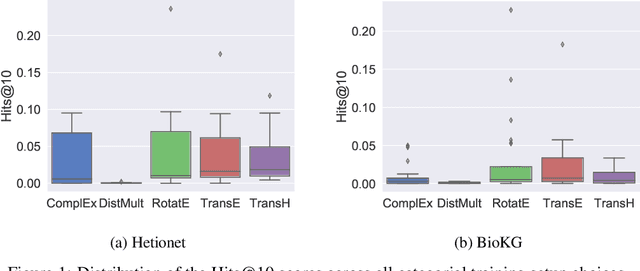
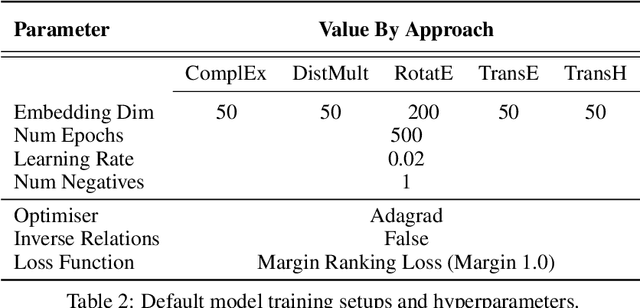
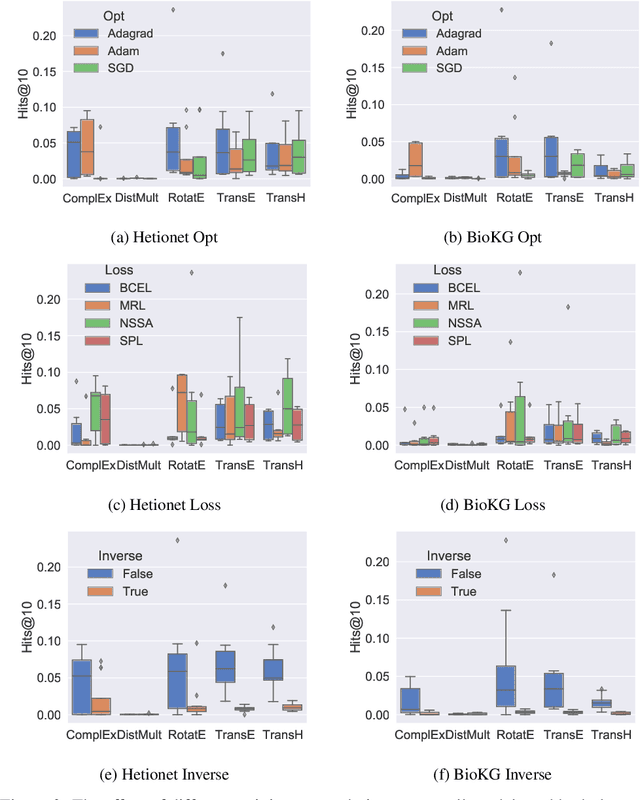
Knowledge Graphs (KG) and associated Knowledge Graph Embedding (KGE) models have recently begun to be explored in the context of drug discovery and have the potential to assist in key challenges such as target identification. In the drug discovery domain, KGs can be employed as part of a process which can result in lab-based experiments being performed, or impact on other decisions, incurring significant time and financial costs and most importantly, ultimately influencing patient healthcare. For KGE models to have impact in this domain, a better understanding of not only of performance, but also the various factors which determine it, is required. In this study we investigate, over the course of many thousands of experiments, the predictive performance of five KGE models on two public drug discovery-oriented KGs. Our goal is not to focus on the best overall model or configuration, instead we take a deeper look at how performance can be affected by changes in the training setup, choice of hyperparameters, model parameter initialisation seed and different splits of the datasets. Our results highlight that these factors have significant impact on performance and can even affect the ranking of models. Indeed these factors should be reported along with model architectures to ensure complete reproducibility and fair comparisons of future work, and we argue this is critical for the acceptance of use, and impact of KGEs in a biomedical setting. To aid reproducibility of our own work, we release all experimentation code.
Predicting Potential Drug Targets Using Tensor Factorisation and Knowledge Graph Embeddings
May 20, 2021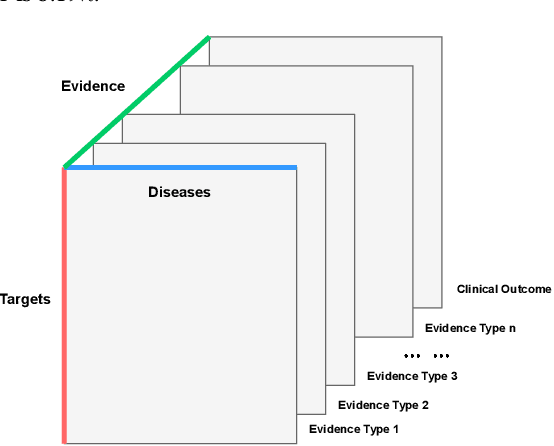
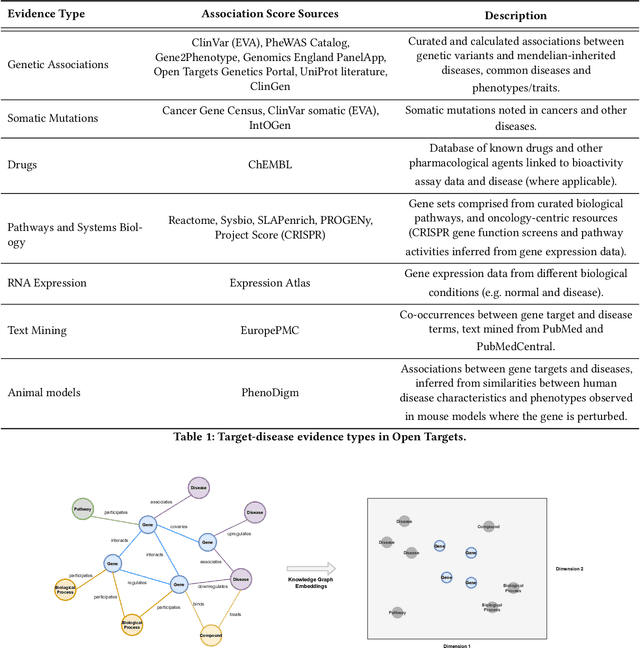
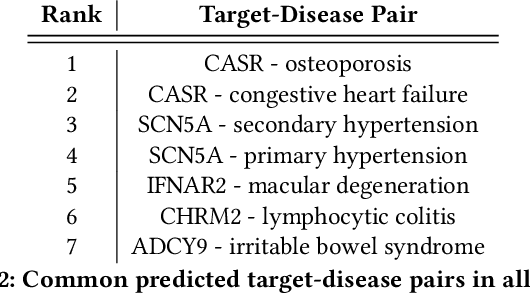
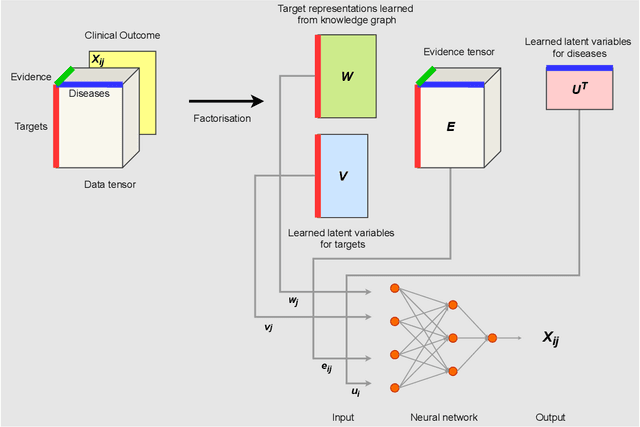
The drug discovery and development process is a long and expensive one, costing over 1 billion USD on average per drug and taking 10-15 years. To reduce the high levels of attrition throughout the process, there has been a growing interest in applying machine learning methodologies to various stages of drug discovery process in the recent decade, including at the earliest stage - identification of druggable disease genes. In this paper, we have developed a new tensor factorisation model to predict potential drug targets (i.e.,genes or proteins) for diseases. We created a three dimensional tensor which consists of 1,048 targets, 860 diseases and 230,011 evidence attributes and clinical outcomes connecting them, using data extracted from the Open Targets and PharmaProjects databases. We enriched the data with gene representations learned from a drug discovery-oriented knowledge graph and applied our proposed method to predict the clinical outcomes for unseen target and dis-ease pairs. We designed three evaluation strategies to measure the prediction performance and benchmarked several commonly used machine learning classifiers together with matrix and tensor factorisation methods. The result shows that incorporating knowledge graph embeddings significantly improves the prediction accuracy and that training tensor factorisation alongside a dense neural network outperforms other methods. In summary, our framework combines two actively studied machine learning approaches to disease target identification, tensor factorisation and knowledge graph representation learning, which could be a promising avenue for further exploration in data-driven drug discovery.
A Review of Biomedical Datasets Relating to Drug Discovery: A Knowledge Graph Perspective
Feb 26, 2021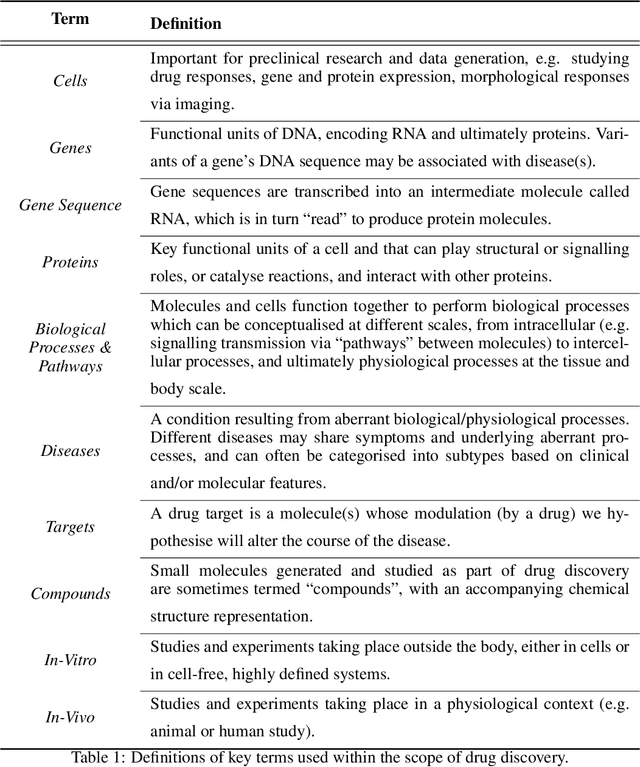
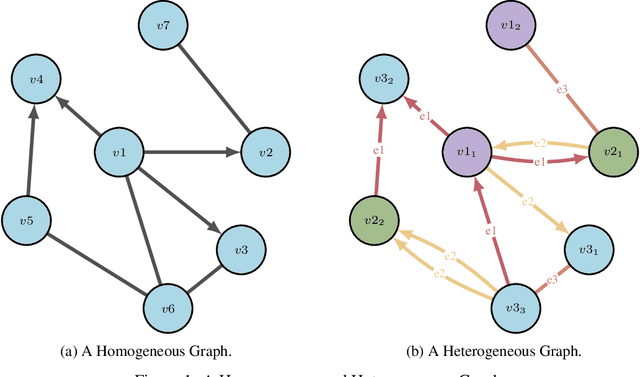
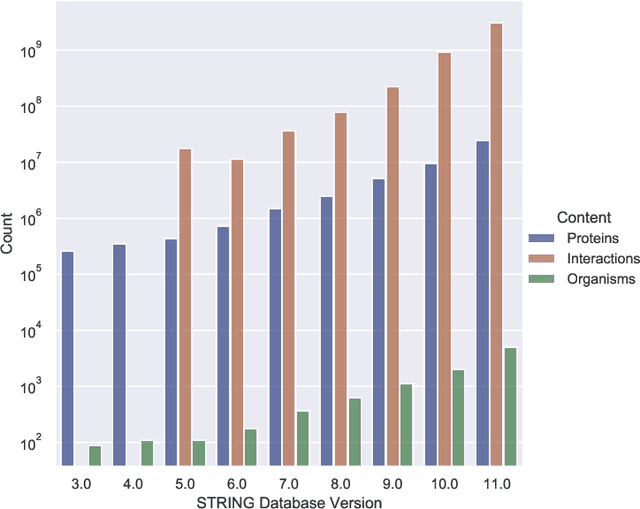
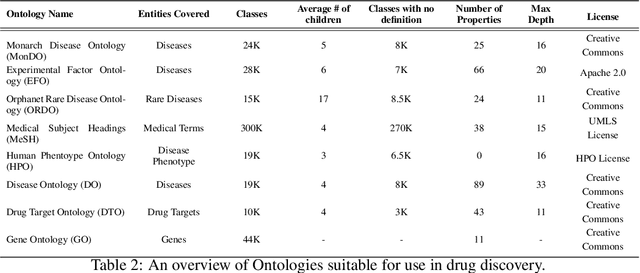
Drug discovery and development is an extremely complex process, with high attrition contributing to the costs of delivering new medicines to patients. Recently, various machine learning approaches have been proposed and investigated to help improve the effectiveness and speed of multiple stages of the drug discovery pipeline. Among these techniques, it is especially those using Knowledge Graphs that are proving to have considerable promise across a range of tasks, including drug repurposing, drug toxicity prediction and target gene-disease prioritisation. In such a knowledge graph-based representation of drug discovery domains, crucial elements including genes, diseases and drugs are represented as entities or vertices, whilst relationships or edges between them indicate some level of interaction. For example, an edge between a disease and drug entity might represent a successful clinical trial, or an edge between two drug entities could indicate a potentially harmful interaction. In order to construct high-quality and ultimately informative knowledge graphs however, suitable data and information is of course required. In this review, we detail publicly available primary data sources containing information suitable for use in constructing various drug discovery focused knowledge graphs. We aim to help guide machine learning and knowledge graph practitioners who are interested in applying new techniques to the drug discovery field, but who may be unfamiliar with the relevant data sources. Overall we hope this review will help motivate more machine learning researchers to explore combining knowledge graphs and machine learning to help solve key and emerging questions in the drug discovery domain.
Not Half Bad: Exploring Half-Precision in Graph Convolutional Neural Networks
Oct 23, 2020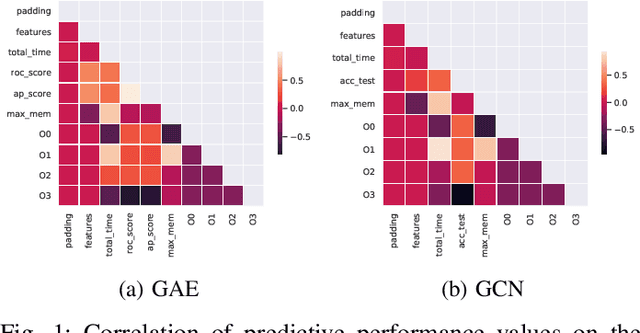
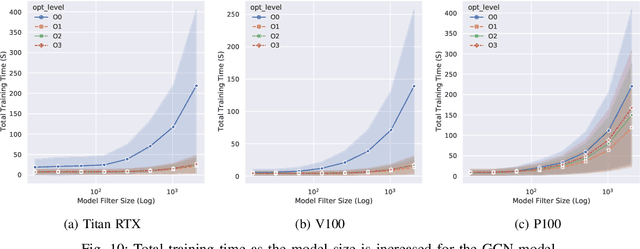
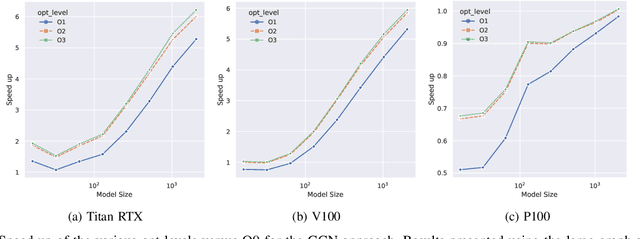
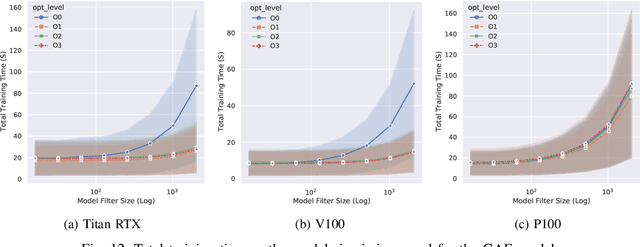
With the growing significance of graphs as an effective representation of data in numerous applications, efficient graph analysis using modern machine learning is receiving a growing level of attention. Deep learning approaches often operate over the entire adjacency matrix -- as the input and intermediate network layers are all designed in proportion to the size of the adjacency matrix -- leading to intensive computation and large memory requirements as the graph size increases. It is therefore desirable to identify efficient measures to reduce both run-time and memory requirements allowing for the analysis of the largest graphs possible. The use of reduced precision operations within the forward and backward passes of a deep neural network along with novel specialised hardware in modern GPUs can offer promising avenues towards efficiency. In this paper, we provide an in-depth exploration of the use of reduced-precision operations, easily integrable into the highly popular PyTorch framework, and an analysis of the effects of Tensor Cores on graph convolutional neural networks. We perform an extensive experimental evaluation of three GPU architectures and two widely-used graph analysis tasks (vertex classification and link prediction) using well-known benchmark and synthetically generated datasets. Thus allowing us to make important observations on the effects of reduced-precision operations and Tensor Cores on computational and memory usage of graph convolutional neural networks -- often neglected in the literature.
Rank over Class: The Untapped Potential of Ranking in Natural Language Processing
Sep 20, 2020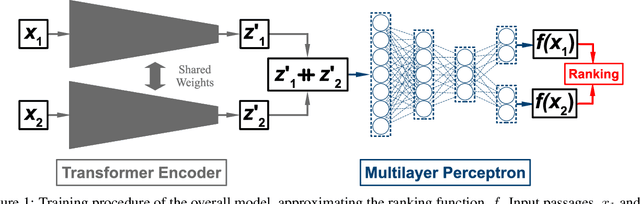
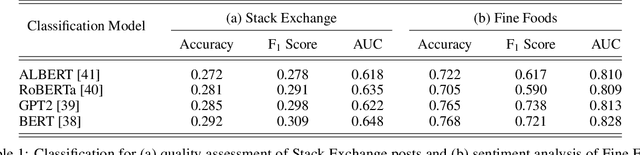
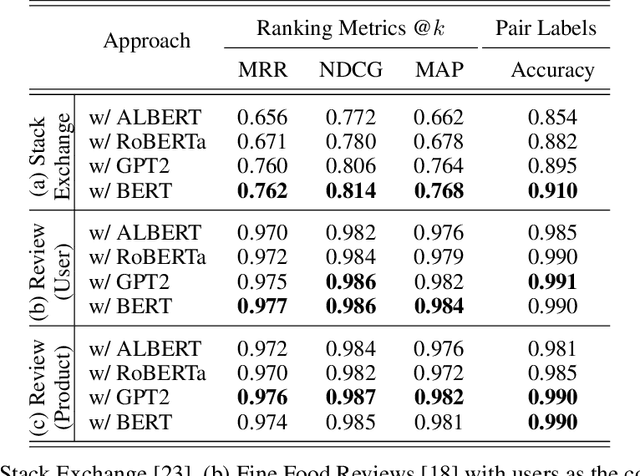
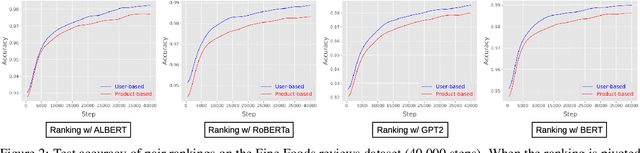
Text classification has long been a staple in natural language processing with applications spanning across sentiment analysis, online content tagging, recommender systems and spam detection. However, text classification, by nature, suffers from a variety of issues stemming from dataset imbalance, text ambiguity, subjectivity and the lack of linguistic context in the data. In this paper, we explore the use of text ranking, commonly used in information retrieval, to carry out challenging classification-based tasks. We propose a novel end-to-end ranking approach consisting of a Transformer network responsible for producing representations for a pair of text sequences, which are in turn passed into a context aggregating network outputting ranking scores used to determine an ordering to the sequences based on some notion of relevance. We perform numerous experiments on publicly-available datasets and investigate the possibility of applying our ranking approach to certain problems often addressed using classification. In an experiment on a heavily-skewed sentiment analysis dataset, converting ranking results to classification labels yields an approximately 22% improvement over state-of-the-art text classification, demonstrating the efficacy of text ranking over text classification in certain scenarios.
BLOB : A Probabilistic Model for Recommendation that Combines Organic and Bandit Signals
Aug 28, 2020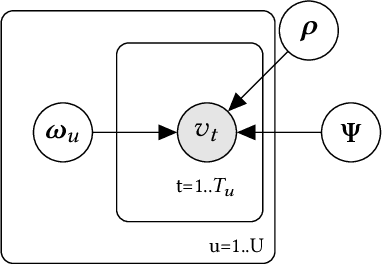
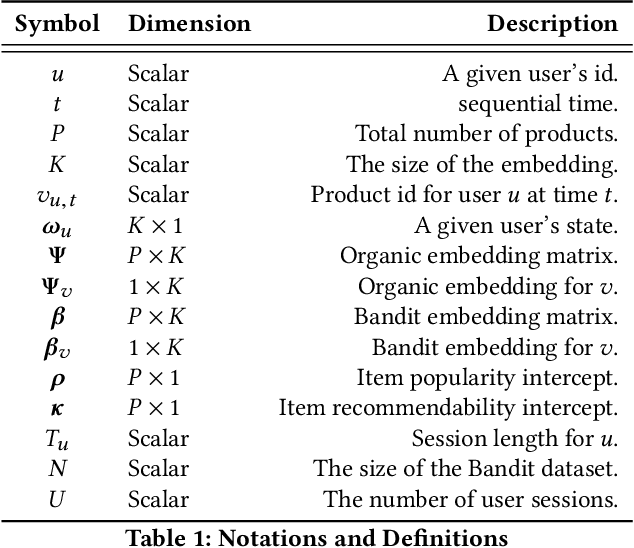
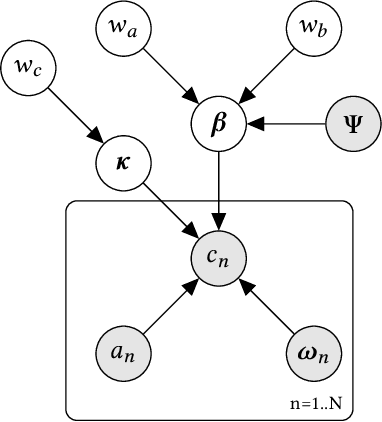
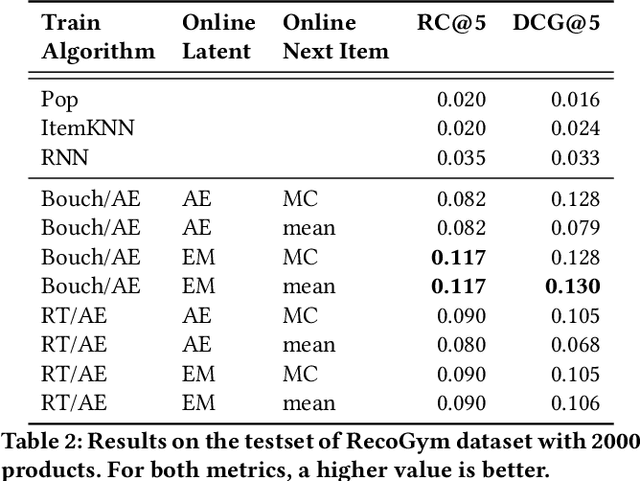
A common task for recommender systems is to build a pro le of the interests of a user from items in their browsing history and later to recommend items to the user from the same catalog. The users' behavior consists of two parts: the sequence of items that they viewed without intervention (the organic part) and the sequences of items recommended to them and their outcome (the bandit part). In this paper, we propose Bayesian Latent Organic Bandit model (BLOB), a probabilistic approach to combine the 'or-ganic' and 'bandit' signals in order to improve the estimation of recommendation quality. The bandit signal is valuable as it gives direct feedback of recommendation performance, but the signal quality is very uneven, as it is highly concentrated on the recommendations deemed optimal by the past version of the recom-mender system. In contrast, the organic signal is typically strong and covers most items, but is not always relevant to the recommendation task. In order to leverage the organic signal to e ciently learn the bandit signal in a Bayesian model we identify three fundamental types of distances, namely action-history, action-action and history-history distances. We implement a scalable approximation of the full model using variational auto-encoders and the local re-paramerization trick. We show using extensive simulation studies that our method out-performs or matches the value of both state-of-the-art organic-based recommendation algorithms, and of bandit-based methods (both value and policy-based) both in organic and bandit-rich environments.
Camera Bias in a Fine Grained Classification Task
Jul 16, 2020
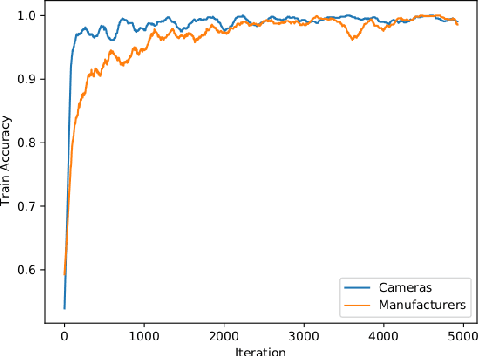
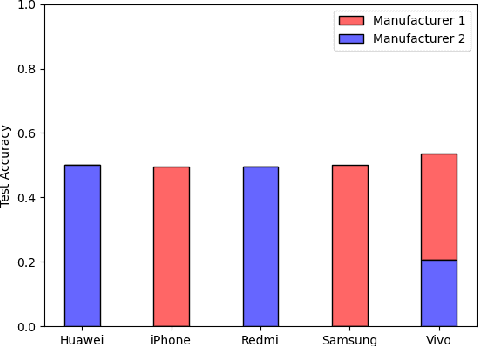
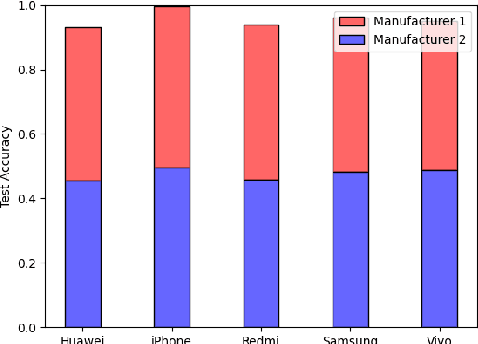
We show that correlations between the camera used to acquire an image and the class label of that image can be exploited by convolutional neural networks (CNN), resulting in a model that "cheats" at an image classification task by recognizing which camera took the image and inferring the class label from the camera. We show that models trained on a dataset with camera / label correlations do not generalize well to images in which those correlations are absent, nor to images from unencountered cameras. Furthermore, we investigate which visual features they are exploiting for camera recognition. Our experiments present evidence against the importance of global color statistics, lens deformation and chromatic aberration, and in favor of high frequency features, which may be introduced by image processing algorithms built into the cameras.