 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient and Equivariant Graph Networks for Predicting Quantum Hamiltonian
Jun 08, 2023



We consider the prediction of the Hamiltonian matrix, which finds use in quantum chemistry and condensed matter physics. Efficiency and equivariance are two important, but conflicting factors. In this work, we propose a SE(3)-equivariant network, named QHNet, that achieves efficiency and equivariance. Our key advance lies at the innovative design of QHNet architecture, which not only obeys the underlying symmetries, but also enables the reduction of number of tensor products by 92\%. In addition, QHNet prevents the exponential growth of channel dimension when more atom types are involved. We perform experiments on MD17 datasets, including four molecular systems. Experimental results show that our QHNet can achieve comparable performance to the state of the art methods at a significantly faster speed. Besides, our QHNet consumes 50\% less memory due to its streamlined architecture. Our code is publicly available as part of the AIRS library (\url{https://github.com/divelab/AIRS}).
A Score-Based Model for Learning Neural Wavefunctions
May 25, 2023



Quantum Monte Carlo coupled with neural network wavefunctions has shown success in computing ground states of quantum many-body systems. Existing optimization approaches compute the energy by sampling local energy from an explicit probability distribution given by the wavefunction. In this work, we provide a new optimization framework for obtaining properties of quantum many-body ground states using score-based neural networks. Our new framework does not require explicit probability distribution and performs the sampling via Langevin dynamics. Our method is based on the key observation that the local energy is directly related to scores, defined as the gradient of the logarithmic wavefunction. Inspired by the score matching and diffusion Monte Carlo methods, we derive a weighted score matching objective to guide our score-based models to converge correctly to ground states. We first evaluate our approach with experiments on quantum harmonic traps, and results show that it can accurately learn ground states of atomic systems. By implicitly modeling high-dimensional data distributions, our work paves the way toward a more efficient representation of quantum systems.
A Latent Diffusion Model for Protein Structure Generation
May 06, 2023



Proteins are complex biomolecules that perform a variety of crucial functions within living organisms. Designing and generating novel proteins can pave the way for many future synthetic biology applications, including drug discovery. However, it remains a challenging computational task due to the large modeling space of protein structures. In this study, we propose a latent diffusion model that can reduce the complexity of protein modeling while flexibly capturing the distribution of natural protein structures in a condensed latent space. Specifically, we propose an equivariant protein autoencoder that embeds proteins into a latent space and then uses an equivariant diffusion model to learn the distribution of the latent protein representations. Experimental results demonstrate that our method can effectively generate novel protein backbone structures with high designability and efficiency.
A new perspective on building efficient and expressive 3D equivariant graph neural networks
Apr 07, 2023Geometric deep learning enables the encoding of physical symmetries in modeling 3D objects. Despite rapid progress in encoding 3D symmetries into Graph Neural Networks (GNNs), a comprehensive evaluation of the expressiveness of these networks through a local-to-global analysis lacks today. In this paper, we propose a local hierarchy of 3D isomorphism to evaluate the expressive power of equivariant GNNs and investigate the process of representing global geometric information from local patches. Our work leads to two crucial modules for designing expressive and efficient geometric GNNs; namely local substructure encoding (LSE) and frame transition encoding (FTE). To demonstrate the applicability of our theory, we propose LEFTNet which effectively implements these modules and achieves state-of-the-art performance on both scalar-valued and vector-valued molecular property prediction tasks. We further point out the design space for future developments of equivariant graph neural networks. Our codes are available at \url{https://github.com/yuanqidu/LeftNet}.
Provably Convergent Subgraph-wise Sampling for Fast GNN Training
Mar 17, 2023



Subgraph-wise sampling -- a promising class of mini-batch training techniques for graph neural networks (GNNs -- is critical for real-world applications. During the message passing (MP) in GNNs, subgraph-wise sampling methods discard messages outside the mini-batches in backward passes to avoid the well-known neighbor explosion problem, i.e., the exponentially increasing dependencies of nodes with the number of MP iterations. However, discarding messages may sacrifice the gradient estimation accuracy, posing significant challenges to their convergence analysis and convergence speeds. To address this challenge, we propose a novel subgraph-wise sampling method with a convergence guarantee, namely Local Message Compensation (LMC). To the best of our knowledge, LMC is the first subgraph-wise sampling method with provable convergence. The key idea is to retrieve the discarded messages in backward passes based on a message passing formulation of backward passes. By efficient and effective compensations for the discarded messages in both forward and backward passes, LMC computes accurate mini-batch gradients and thus accelerates convergence. Moreover, LMC is applicable to various MP-based GNN architectures, including convolutional GNNs (finite message passing iterations with different layers) and recurrent GNNs (infinite message passing iterations with a shared layer). Experiments on large-scale benchmarks demonstrate that LMC is significantly faster than state-of-the-art subgraph-wise sampling methods.
Generalization in Visual Reinforcement Learning with the Reward Sequence Distribution
Feb 19, 2023Generalization in partially observed markov decision processes (POMDPs) is critical for successful applications of visual reinforcement learning (VRL) in real scenarios. A widely used idea is to learn task-relevant representations that encode task-relevant information of common features in POMDPs, i.e., rewards and transition dynamics. As transition dynamics in the latent state space -- which are task-relevant and invariant to visual distractions -- are unknown to the agents, existing methods alternatively use transition dynamics in the observation space to extract task-relevant information in transition dynamics. However, such transition dynamics in the observation space involve task-irrelevant visual distractions, degrading the generalization performance of VRL methods. To tackle this problem, we propose the reward sequence distribution conditioned on the starting observation and the predefined subsequent action sequence (RSD-OA). The appealing features of RSD-OA include that: (1) RSD-OA is invariant to visual distractions, as it is conditioned on the predefined subsequent action sequence without task-irrelevant information from transition dynamics, and (2) the reward sequence captures long-term task-relevant information in both rewards and transition dynamics. Experiments demonstrate that our representation learning approach based on RSD-OA significantly improves the generalization performance on unseen environments, outperforming several state-of-the-arts on DeepMind Control tasks with visual distractions.
DiffBP: Generative Diffusion of 3D Molecules for Target Protein Binding
Dec 01, 2022



Generating molecules that bind to specific proteins is an important but challenging task in drug discovery. Previous works usually generate atoms in an auto-regressive way, where element types and 3D coordinates of atoms are generated one by one. However, in real-world molecular systems, the interactions among atoms in an entire molecule are global, leading to the energy function pair-coupled among atoms. With such energy-based consideration, the modeling of probability should be based on joint distributions, rather than sequentially conditional ones. Thus, the unnatural sequentially auto-regressive modeling of molecule generation is likely to violate the physical rules, thus resulting in poor properties of the generated molecules. In this work, a generative diffusion model for molecular 3D structures based on target proteins as contextual constraints is established, at a full-atom level in a non-autoregressive way. Given a designated 3D protein binding site, our model learns the generative process that denoises both element types and 3D coordinates of an entire molecule, with an equivariant network. Experimentally, the proposed method shows competitive performance compared with prevailing works in terms of high affinity with proteins and appropriate molecule sizes as well as other drug properties such as drug-likeness of the generated molecules.
Gradient-Guided Importance Sampling for Learning Binary Energy-Based Models
Oct 11, 2022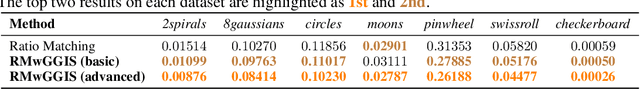

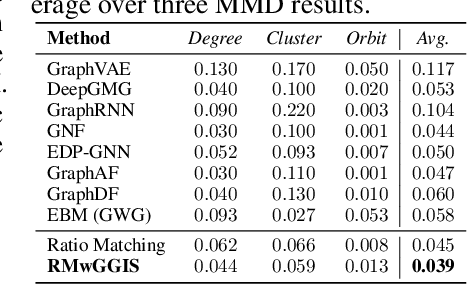
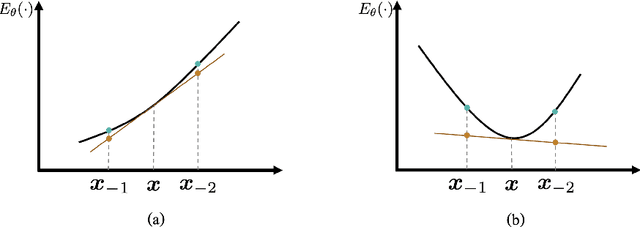
Learning energy-based models (EBMs) is known to be difficult especially on discrete data where gradient-based learning strategies cannot be applied directly. Although ratio matching is a sound method to learn discrete EBMs, it suffers from expensive computation and excessive memory requirement, thereby resulting in difficulties for learning EBMs on high-dimensional data. Motivated from these limitations, in this study, we propose ratio matching with gradient-guided importance sampling (RMwGGIS). Particularly, we use the gradient of the energy function w.r.t. the discrete data space to approximately construct the provably optimal proposal distribution, which is subsequently used by importance sampling to efficiently estimate the original ratio matching objective. We perform experiments on density modeling over synthetic discrete data, graph generation, and training Ising models to evaluate our proposed method. The experimental results demonstrate that our method can significantly alleviate the limitations of ratio matching, perform more effectively in practice, and scale to high-dimensional problems. Our implementation is available at {https://github.com/divelab/RMwGGIS.
Periodic Graph Transformers for Crystal Material Property Prediction
Sep 23, 2022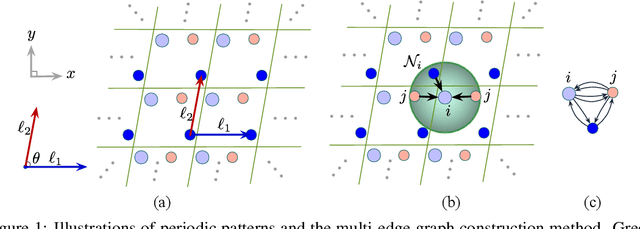
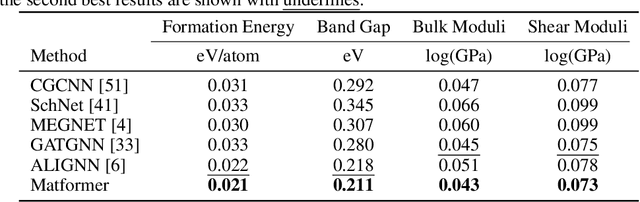
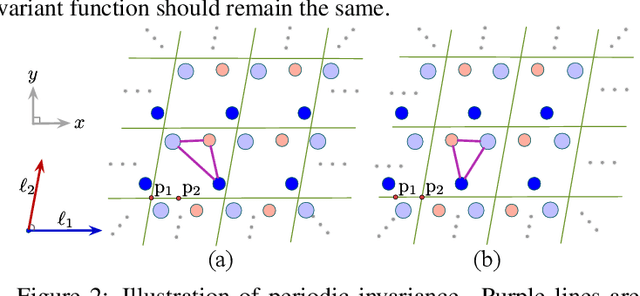
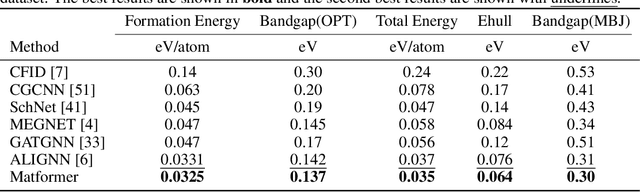
We consider representation learning on periodic graphs encoding crystal materials. Different from regular graphs, periodic graphs consist of a minimum unit cell repeating itself on a regular lattice in 3D space. How to effectively encode these periodic structures poses unique challenges not present in regular graph representation learning. In addition to being E(3) invariant, periodic graph representations need to be periodic invariant. That is, the learned representations should be invariant to shifts of cell boundaries as they are artificially imposed. Furthermore, the periodic repeating patterns need to be captured explicitly as lattices of different sizes and orientations may correspond to different materials. In this work, we propose a transformer architecture, known as Matformer, for periodic graph representation learning. Our Matformer is designed to be invariant to periodicity and can capture repeating patterns explicitly. In particular, Matformer encodes periodic patterns by efficient use of geometric distances between the same atoms in neighboring cells. Experimental results on multiple common benchmark datasets show that our Matformer outperforms baseline methods consistently. In addition, our results demonstrate the importance of periodic invariance and explicit repeating pattern encoding for crystal representation learning.
Learning Protein Representations via Complete 3D Graph Networks
Jul 26, 2022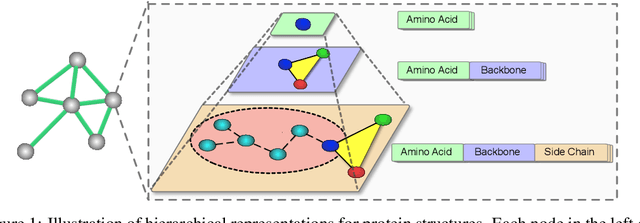
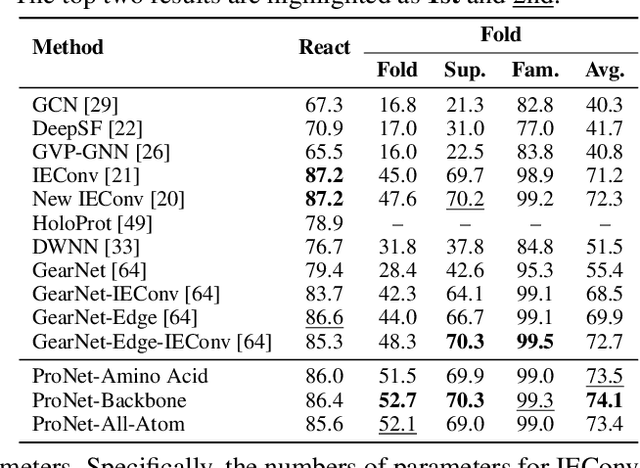
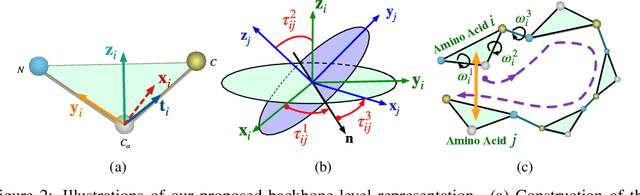
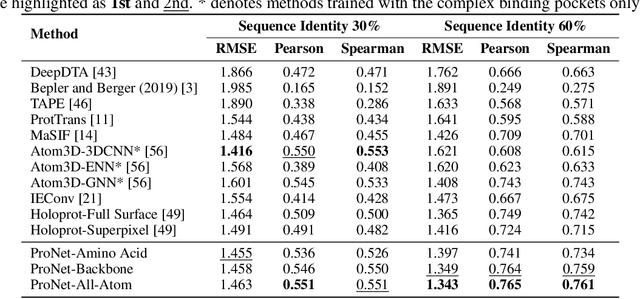
We consider representation learning for proteins with 3D structures. We build 3D graphs based on protein structures and develop graph networks to learn their representations. Depending on the levels of details that we wish to capture, protein representations can be computed at different levels, \emph{e.g.}, the amino acid, backbone, or all-atom levels. Importantly, there exist hierarchical relations among different levels. In this work, we propose to develop a novel hierarchical graph network, known as ProNet, to capture the relations. Our ProNet is very flexible and can be used to compute protein representations at different levels of granularity. We show that, given a base 3D graph network that is complete, our ProNet representations are also complete at all levels. To close the loop, we develop a complete and efficient 3D graph network to be used as a base model, making our ProNet complete. We conduct experiments on multiple downstream tasks. Results show that ProNet outperforms recent methods on most datasets. In addition, results indicate that different downstream tasks may require representations at different levels. Our code is available as part of the DIG library (\url{https://github.com/divelab/DIG}).