 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMitosis Detection in the Wild: Multi-Tumor and Context-Aware Generalization in the MIDOG 2025 Challenge
Jun 05, 2026Automated mitosis detection is a well-established task in computational pathology. While previous benchmarks focused on scanner-induced domain shift, clinical "real-world" application requires models to be robust across the vast variance to be expected in the histological landscape. The MItosis DOmain Generalization (MIDOG) 2025 challenge was designed to evaluate algorithmic performance across unprecedented biological and contextual diversity. We curated a test dataset of 365 cases, encompassing 12 distinct human, canine and feline tumor types, digitized across multiple scanning platforms. Moving beyond hand-selected hotspots, the challenge required detection also in random tissue areas (representative of the whole slide detection situation) and challenging areas (areas rich in hard negatives). In the second track, we introduced the classification of atypical mitotic figures (AMFs). There were 18 teams submitting to the detection track, with F1 scores ranging up to 0.740. In the AMF detection track, we had 21 submissions with balanced accuracy values up to 0.908. Our analysis reveals that while most models perform reliably in traditional hotspots, significant performance degradation occurs in challenging ROIs, where false positive rates tripled. Furthermore, performance varied significantly across the 12 tumor types, highlighting "blind spots" in current state-of-the-art architectures when encountering rare or highly pleomorphic malignancies. Moreover, we evaluated the effectiveness of ensembling and found a mean increases of 1.5 and 1.3 percentage points in F1 score and balanced accuracy, respectively. In contrast, TTA showed no relevant improvement. MIDOG 2025 demonstrates that "in the wild" mitosis detection remains a significant hurdle. The transition from hotspot-only evaluation to a multi-contextual framework provides a more realistic proxy for clinical reliability.
From Traditional to Deep Learning Approaches in Whole Slide Image Registration: A Methodological Review
Feb 26, 2025



Whole slide image (WSI) registration is an essential task for analysing the tumour microenvironment (TME) in histopathology. It involves the alignment of spatial information between WSIs of the same section or serial sections of a tissue sample. The tissue sections are usually stained with single or multiple biomarkers before imaging, and the goal is to identify neighbouring nuclei along the Z-axis for creating a 3D image or identifying subclasses of cells in the TME. This task is considerably more challenging compared to radiology image registration, such as magnetic resonance imaging or computed tomography, due to various factors. These include gigapixel size of images, variations in appearance between differently stained tissues, changes in structure and morphology between non-consecutive sections, and the presence of artefacts, tears, and deformations. Currently, there is a noticeable gap in the literature regarding a review of the current approaches and their limitations, as well as the challenges and opportunities they present. We aim to provide a comprehensive understanding of the available approaches and their application for various purposes. Furthermore, we investigate current deep learning methods used for WSI registration, emphasising their diverse methodologies. We examine the available datasets and explore tools and software employed in the field. Finally, we identify open challenges and potential future trends in this area of research.
Benchmarking Domain Generalization Algorithms in Computational Pathology
Sep 25, 2024



Deep learning models have shown immense promise in computational pathology (CPath) tasks, but their performance often suffers when applied to unseen data due to domain shifts. Addressing this requires domain generalization (DG) algorithms. However, a systematic evaluation of DG algorithms in the CPath context is lacking. This study aims to benchmark the effectiveness of 30 DG algorithms on 3 CPath tasks of varying difficulty through 7,560 cross-validation runs. We evaluate these algorithms using a unified and robust platform, incorporating modality-specific techniques and recent advances like pretrained foundation models. Our extensive cross-validation experiments provide insights into the relative performance of various DG strategies. We observe that self-supervised learning and stain augmentation consistently outperform other methods, highlighting the potential of pretrained models and data augmentation. Furthermore, we introduce a new pan-cancer tumor detection dataset (HISTOPANTUM) as a benchmark for future research. This study offers valuable guidance to researchers in selecting appropriate DG approaches for CPath tasks.
TIAViz: A Browser-based Visualization Tool for Computational Pathology Models
Feb 15, 2024


Digital pathology has gained significant traction in modern healthcare systems. This shift from optical microscopes to digital imagery brings with it the potential for improved diagnosis, efficiency, and the integration of AI tools into the pathologists workflow. A critical aspect of this is visualization. Throughout the development of a machine learning (ML) model in digital pathology, it is crucial to have flexible, openly available tools to visualize models, from their outputs and predictions to the underlying annotations and images used to train or test a model. We introduce TIAViz, a Python-based visualization tool built into TIAToolbox which allows flexible, interactive, fully zoomable overlay of a wide variety of information onto whole slide images, including graphs, heatmaps, segmentations, annotations and other WSIs. The UI is browser-based, allowing use either locally, on a remote machine, or on a server to provide publicly available demos. This tool is open source and is made available at: https://github.com/TissueImageAnalytics/tiatoolbox and via pip installation (pip install tiatoolbox) and conda as part of TIAToolbox.
An Automated Pipeline for Tumour-Infiltrating Lymphocyte Scoring in Breast Cancer
Nov 21, 2023Tumour-infiltrating lymphocytes (TILs) are considered as a valuable prognostic markers in both triple-negative and human epidermal growth factor receptor 2 (HER2) positive breast cancer. In this study, we introduce an innovative deep learning pipeline based on the Efficient-UNet architecture to predict the TILs score for breast cancer whole-slide images (WSIs). We first segment tumour and stromal regions in order to compute a tumour bulk mask. We then detect TILs within the tumour-associated stroma, generating a TILs score by closely mirroring the pathologist's workflow. Our method exhibits state-of-the-art performance in segmenting tumour/stroma areas and TILs detection, as demonstrated by internal cross-validation on the TiGER Challenge training dataset and evaluation on the final leaderboards. Additionally, our TILs score proves competitive in predicting survival outcomes within the same challenge, underscoring the clinical relevance and potential of our automated TILs scoring pipeline as a breast cancer prognostic tool.
Domain Generalization in Computational Pathology: Survey and Guidelines
Oct 30, 2023



Deep learning models have exhibited exceptional effectiveness in Computational Pathology (CPath) by tackling intricate tasks across an array of histology image analysis applications. Nevertheless, the presence of out-of-distribution data (stemming from a multitude of sources such as disparate imaging devices and diverse tissue preparation methods) can cause \emph{domain shift} (DS). DS decreases the generalization of trained models to unseen datasets with slightly different data distributions, prompting the need for innovative \emph{domain generalization} (DG) solutions. Recognizing the potential of DG methods to significantly influence diagnostic and prognostic models in cancer studies and clinical practice, we present this survey along with guidelines on achieving DG in CPath. We rigorously define various DS types, systematically review and categorize existing DG approaches and resources in CPath, and provide insights into their advantages, limitations, and applicability. We also conduct thorough benchmarking experiments with 28 cutting-edge DG algorithms to address a complex DG problem. Our findings suggest that careful experiment design and CPath-specific Stain Augmentation technique can be very effective. However, there is no one-size-fits-all solution for DG in CPath. Therefore, we establish clear guidelines for detecting and managing DS depending on different scenarios. While most of the concepts, guidelines, and recommendations are given for applications in CPath, we believe that they are applicable to most medical image analysis tasks as well.
Domain generalization across tumor types, laboratories, and species -- insights from the 2022 edition of the Mitosis Domain Generalization Challenge
Sep 27, 2023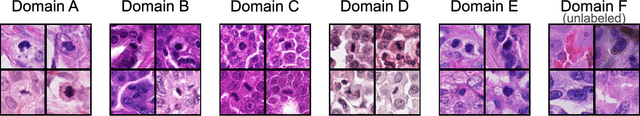
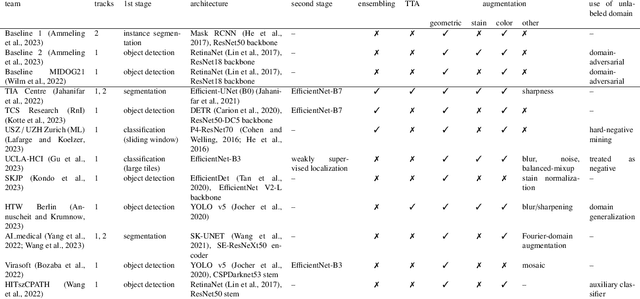
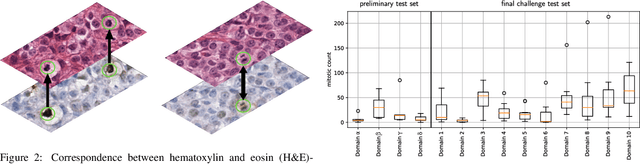
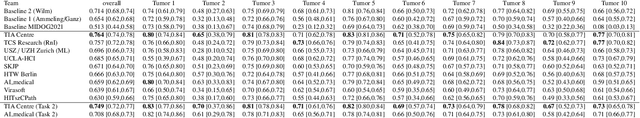
Recognition of mitotic figures in histologic tumor specimens is highly relevant to patient outcome assessment. This task is challenging for algorithms and human experts alike, with deterioration of algorithmic performance under shifts in image representations. Considerable covariate shifts occur when assessment is performed on different tumor types, images are acquired using different digitization devices, or specimens are produced in different laboratories. This observation motivated the inception of the 2022 challenge on MItosis Domain Generalization (MIDOG 2022). The challenge provided annotated histologic tumor images from six different domains and evaluated the algorithmic approaches for mitotic figure detection provided by nine challenge participants on ten independent domains. Ground truth for mitotic figure detection was established in two ways: a three-expert consensus and an independent, immunohistochemistry-assisted set of labels. This work represents an overview of the challenge tasks, the algorithmic strategies employed by the participants, and potential factors contributing to their success. With an $F_1$ score of 0.764 for the top-performing team, we summarize that domain generalization across various tumor domains is possible with today's deep learning-based recognition pipelines. When assessed against the immunohistochemistry-assisted reference standard, all methods resulted in reduced recall scores, but with only minor changes in the order of participants in the ranking.
A Fully Automated and Explainable Algorithm for the Prediction of Malignant Transformation in Oral Epithelial Dysplasia
Jul 06, 2023



Oral epithelial dysplasia (OED) is a premalignant histopathological diagnosis given to lesions of the oral cavity. Its grading suffers from significant inter-/intra- observer variability, and does not reliably predict malignancy progression, potentially leading to suboptimal treatment decisions. To address this, we developed a novel artificial intelligence algorithm that can assign an Oral Malignant Transformation (OMT) risk score, based on histological patterns in the in Haematoxylin and Eosin stained whole slide images, to quantify the risk of OED progression. The algorithm is based on the detection and segmentation of nuclei within (and around) the epithelium using an in-house segmentation model. We then employed a shallow neural network fed with interpretable morphological/spatial features, emulating histological markers. We conducted internal cross-validation on our development cohort (Sheffield; n = 193 cases) followed by independent validation on two external cohorts (Birmingham and Belfast; n = 92 cases). The proposed OMTscore yields an AUROC = 0.74 in predicting whether an OED progresses to malignancy or not. Survival analyses showed the prognostic value of our OMTscore for predicting malignancy transformation, when compared to the manually-assigned WHO and binary grades. Analysis of the correctly predicted cases elucidated the presence of peri-epithelial and epithelium-infiltrating lymphocytes in the most predictive patches of cases that transformed (p < 0.0001). This is the first study to propose a completely automated algorithm for predicting OED transformation based on interpretable nuclear features, whilst being validated on external datasets. The algorithm shows better-than-human-level performance for prediction of OED malignant transformation and offers a promising solution to the challenges of grading OED in routine clinical practice.
Why is the winner the best?
Mar 30, 2023



International benchmarking competitions have become fundamental for the comparative performance assessment of image analysis methods. However, little attention has been given to investigating what can be learnt from these competitions. Do they really generate scientific progress? What are common and successful participation strategies? What makes a solution superior to a competing method? To address this gap in the literature, we performed a multi-center study with all 80 competitions that were conducted in the scope of IEEE ISBI 2021 and MICCAI 2021. Statistical analyses performed based on comprehensive descriptions of the submitted algorithms linked to their rank as well as the underlying participation strategies revealed common characteristics of winning solutions. These typically include the use of multi-task learning (63%) and/or multi-stage pipelines (61%), and a focus on augmentation (100%), image preprocessing (97%), data curation (79%), and postprocessing (66%). The "typical" lead of a winning team is a computer scientist with a doctoral degree, five years of experience in biomedical image analysis, and four years of experience in deep learning. Two core general development strategies stood out for highly-ranked teams: the reflection of the metrics in the method design and the focus on analyzing and handling failure cases. According to the organizers, 43% of the winning algorithms exceeded the state of the art but only 11% completely solved the respective domain problem. The insights of our study could help researchers (1) improve algorithm development strategies when approaching new problems, and (2) focus on open research questions revealed by this work.
CoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.