 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSemiparametric spectral modeling of the Drosophila connectome
May 09, 2017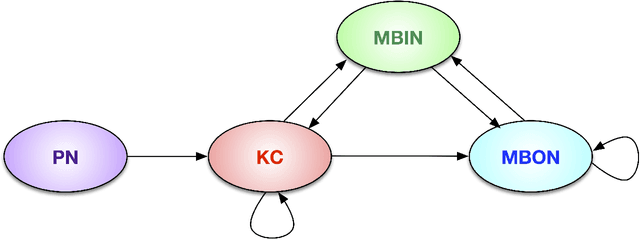
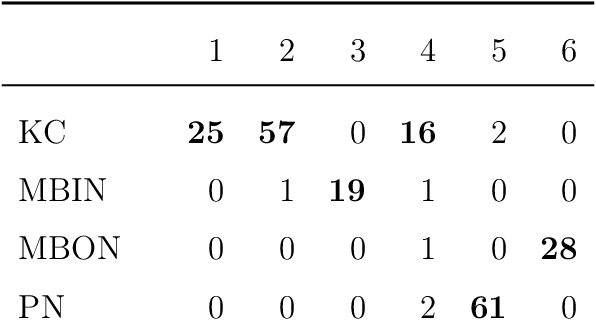
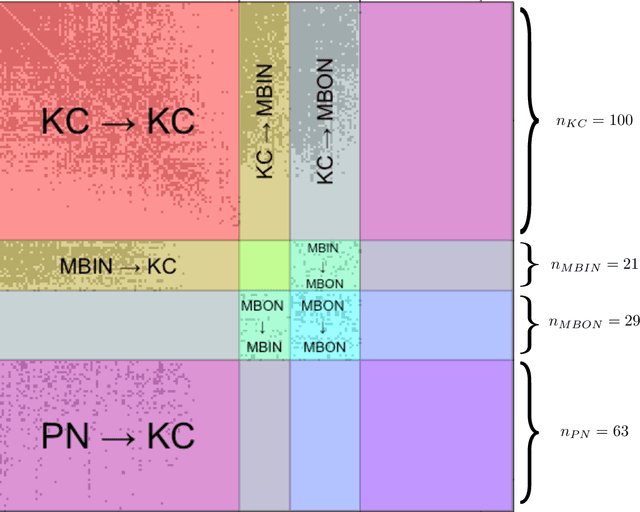

We present semiparametric spectral modeling of the complete larval Drosophila mushroom body connectome. Motivated by a thorough exploratory data analysis of the network via Gaussian mixture modeling (GMM) in the adjacency spectral embedding (ASE) representation space, we introduce the latent structure model (LSM) for network modeling and inference. LSM is a generalization of the stochastic block model (SBM) and a special case of the random dot product graph (RDPG) latent position model, and is amenable to semiparametric GMM in the ASE representation space. The resulting connectome code derived via semiparametric GMM composed with ASE captures latent connectome structure and elucidates biologically relevant neuronal properties.
Manifold Matching using Shortest-Path Distance and Joint Neighborhood Selection
Apr 11, 2017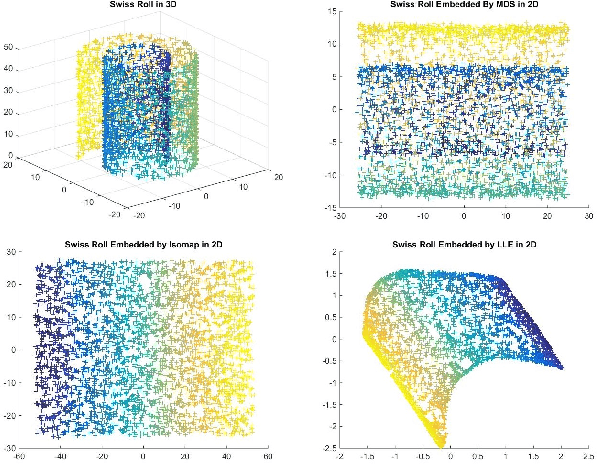
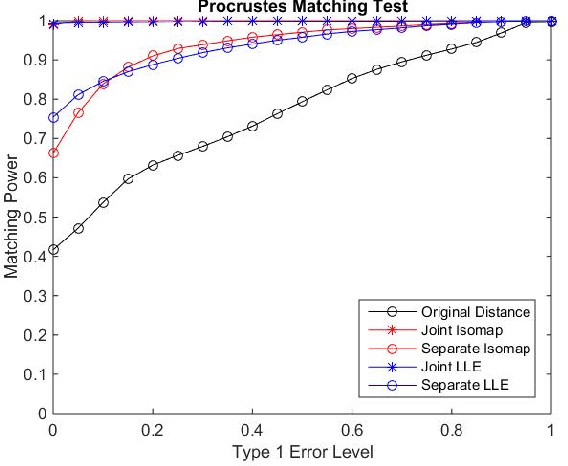
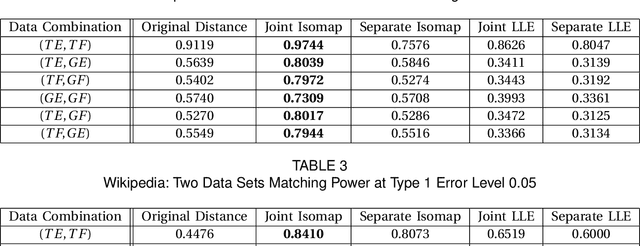
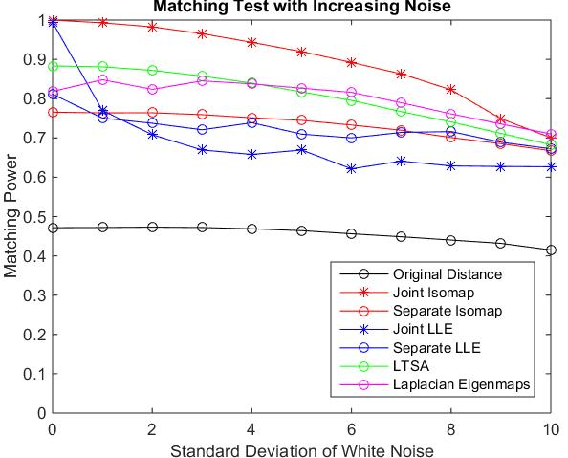
Matching datasets of multiple modalities has become an important task in data analysis. Existing methods often rely on the embedding and transformation of each single modality without utilizing any correspondence information, which often results in sub-optimal matching performance. In this paper, we propose a nonlinear manifold matching algorithm using shortest-path distance and joint neighborhood selection. Specifically, a joint nearest-neighbor graph is built for all modalities. Then the shortest-path distance within each modality is calculated from the joint neighborhood graph, followed by embedding into and matching in a common low-dimensional Euclidean space. Compared to existing algorithms, our approach exhibits superior performance for matching disparate datasets of multiple modalities.
* 13 pages, 8 figures, 2 tables
Joint Embedding of Graphs
Mar 10, 2017

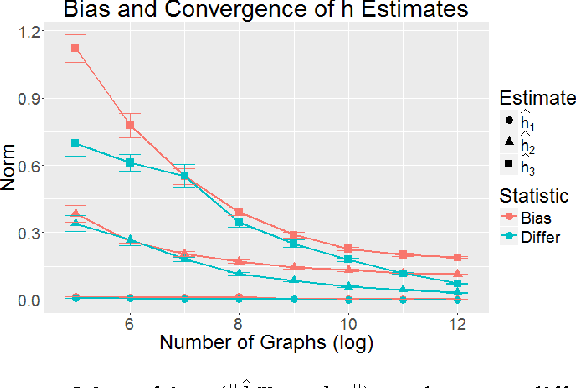
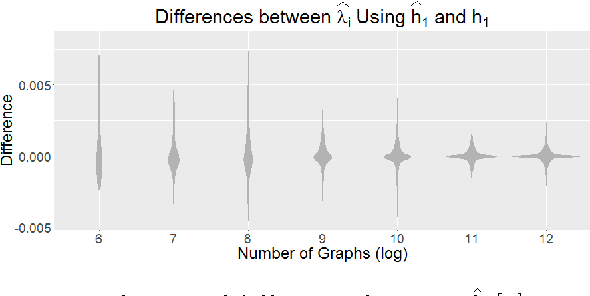
Feature extraction and dimension reduction for networks is critical in a wide variety of domains. Efficiently and accurately learning features for multiple graphs has important applications in statistical inference on graphs. We propose a method to jointly embed multiple undirected graphs. Given a set of graphs, the joint embedding method identifies a linear subspace spanned by rank one symmetric matrices and projects adjacency matrices of graphs into this subspace. The projection coefficients can be treated as features of the graphs. We also propose a random graph model which generalizes classical random graph model and can be used to model multiple graphs. We show through theory and numerical experiments that under the model, the joint embedding method produces estimates of parameters with small errors. Via simulation experiments, we demonstrate that the joint embedding method produces features which lead to state of the art performance in classifying graphs. Applying the joint embedding method to human brain graphs, we find it extract interpretable features that can be used to predict individual composite creativity index.
Probabilistic Fluorescence-Based Synapse Detection
Nov 16, 2016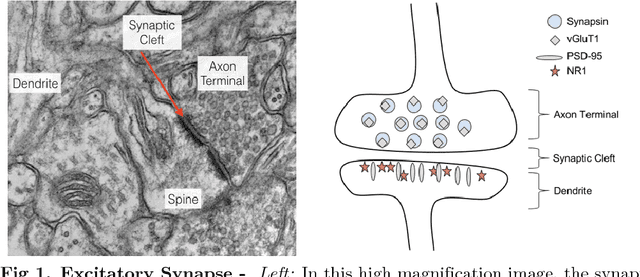


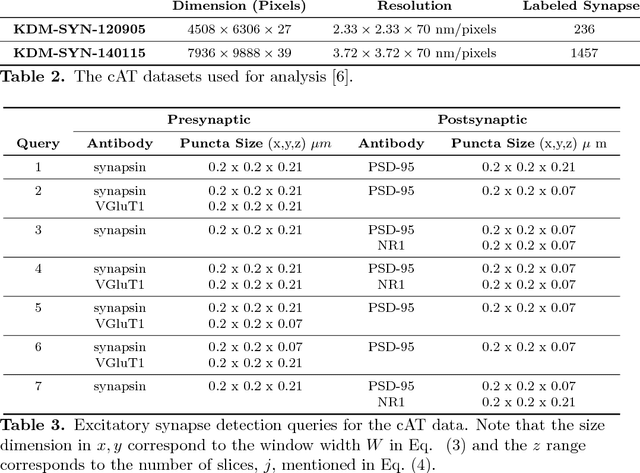
Brain function results from communication between neurons connected by complex synaptic networks. Synapses are themselves highly complex and diverse signaling machines, containing protein products of hundreds of different genes, some in hundreds of copies, arranged in precise lattice at each individual synapse. Synapses are fundamental not only to synaptic network function but also to network development, adaptation, and memory. In addition, abnormalities of synapse numbers or molecular components are implicated in most mental and neurological disorders. Despite their obvious importance, mammalian synapse populations have so far resisted detailed quantitative study. In human brains and most animal nervous systems, synapses are very small and very densely packed: there are approximately 1 billion synapses per cubic millimeter of human cortex. This volumetric density poses very substantial challenges to proteometric analysis at the critical level of the individual synapse. The present work describes new probabilistic image analysis methods for single-synapse analysis of synapse populations in both animal and human brains.
Covariate-assisted spectral clustering
Oct 30, 2016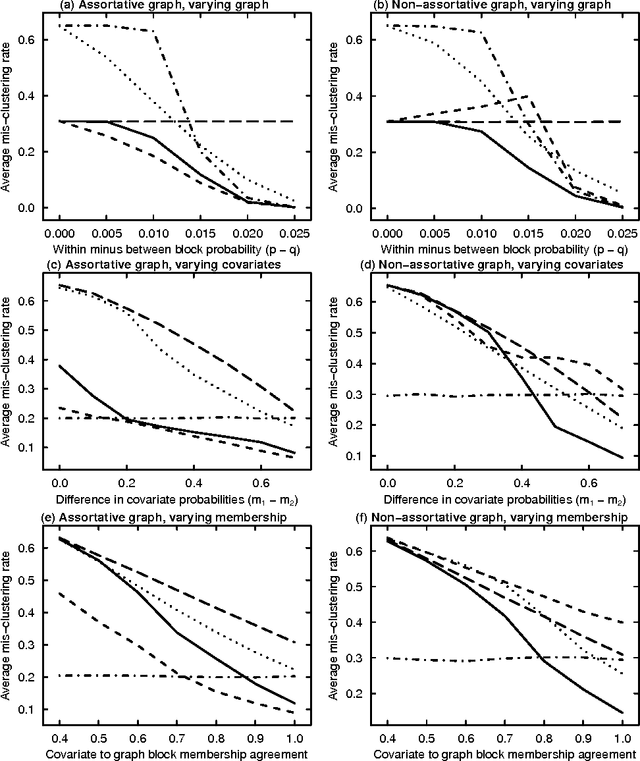
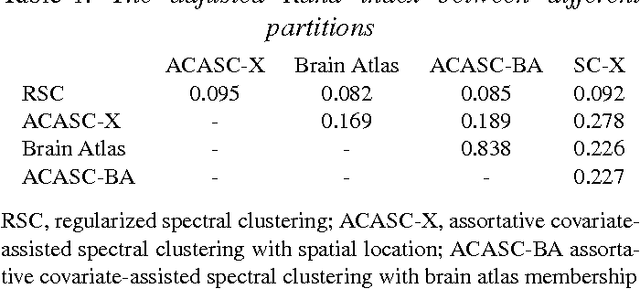
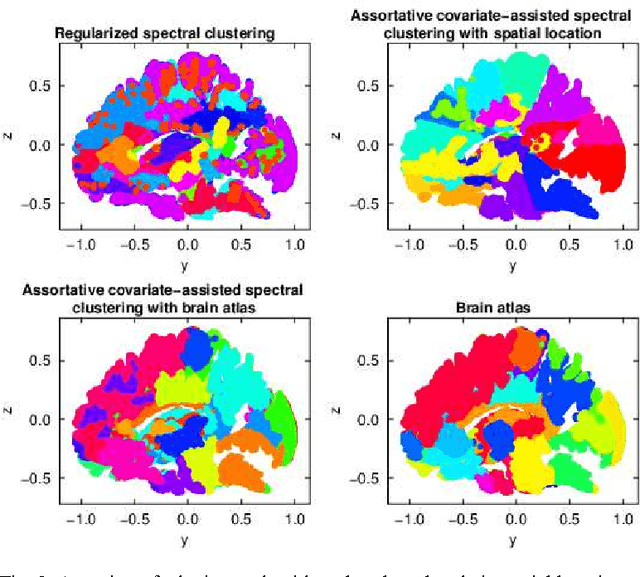
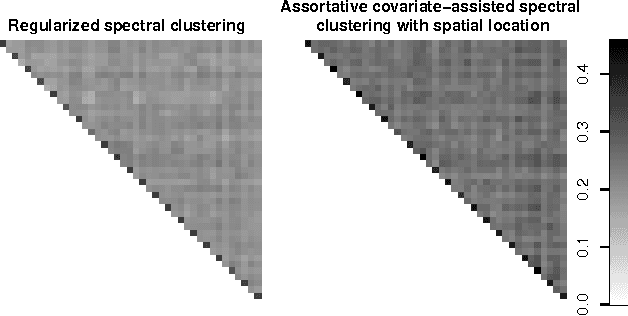
Biological and social systems consist of myriad interacting units. The interactions can be represented in the form of a graph or network. Measurements of these graphs can reveal the underlying structure of these interactions, which provides insight into the systems that generated the graphs. Moreover, in applications such as connectomics, social networks, and genomics, graph data are accompanied by contextualizing measures on each node. We utilize these node covariates to help uncover latent communities in a graph, using a modification of spectral clustering. Statistical guarantees are provided under a joint mixture model that we call the node-contextualized stochastic blockmodel, including a bound on the mis-clustering rate. The bound is used to derive conditions for achieving perfect clustering. For most simulated cases, covariate-assisted spectral clustering yields results superior to regularized spectral clustering without node covariates and to an adaptation of canonical correlation analysis. We apply our clustering method to large brain graphs derived from diffusion MRI data, using the node locations or neurological region membership as covariates. In both cases, covariate-assisted spectral clustering yields clusters that are easier to interpret neurologically.
* 28 pages, 4 figures, includes substantial changes to theoretical results
Quantifying mesoscale neuroanatomy using X-ray microtomography
Jul 26, 2016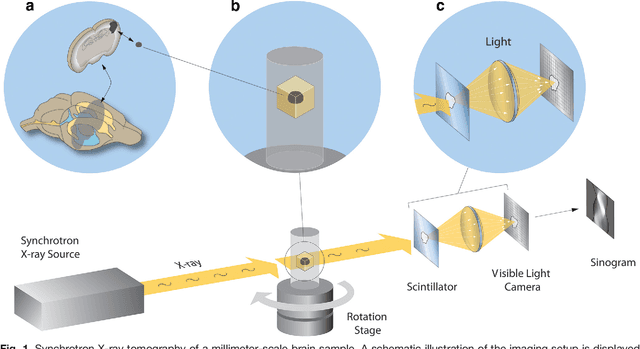
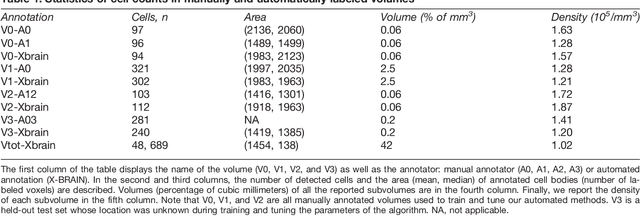
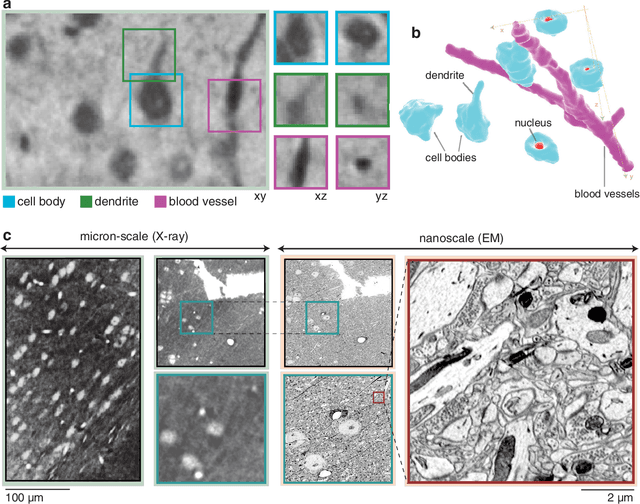
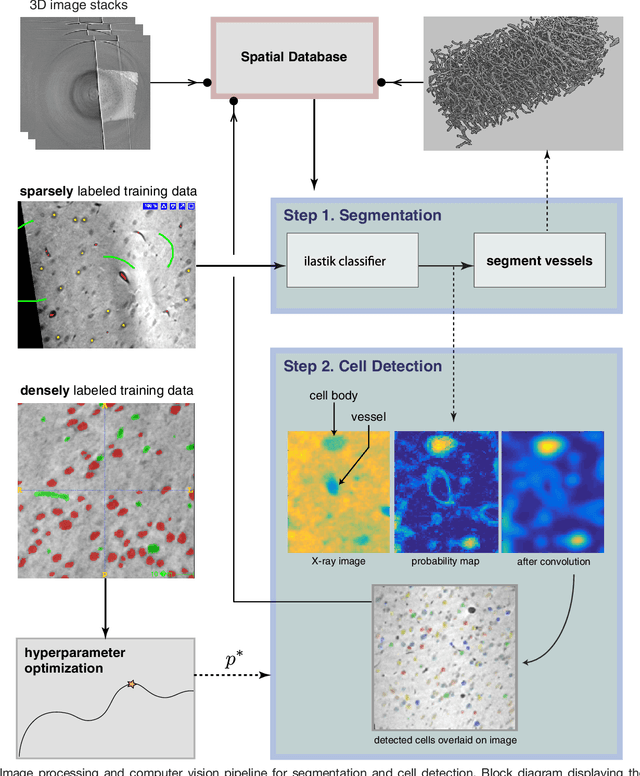
Methods for resolving the 3D microstructure of the brain typically start by thinly slicing and staining the brain, and then imaging each individual section with visible light photons or electrons. In contrast, X-rays can be used to image thick samples, providing a rapid approach for producing large 3D brain maps without sectioning. Here we demonstrate the use of synchrotron X-ray microtomography ($\mu$CT) for producing mesoscale $(1~\mu m^3)$ resolution brain maps from millimeter-scale volumes of mouse brain. We introduce a pipeline for $\mu$CT-based brain mapping that combines methods for sample preparation, imaging, automated segmentation of image volumes into cells and blood vessels, and statistical analysis of the resulting brain structures. Our results demonstrate that X-ray tomography promises rapid quantification of large brain volumes, complementing other brain mapping and connectomics efforts.
Deformably Registering and Annotating Whole CLARITY Brains to an Atlas via Masked LDDMM
May 06, 2016The CLARITY method renders brains optically transparent to enable high-resolution imaging in the structurally intact brain. Anatomically annotating CLARITY brains is necessary for discovering which regions contain signals of interest. Manually annotating whole-brain, terabyte CLARITY images is difficult, time-consuming, subjective, and error-prone. Automatically registering CLARITY images to a pre-annotated brain atlas offers a solution, but is difficult for several reasons. Removal of the brain from the skull and subsequent storage and processing cause variable non-rigid deformations, thus compounding inter-subject anatomical variability. Additionally, the signal in CLARITY images arises from various biochemical contrast agents which only sparsely label brain structures. This sparse labeling challenges the most commonly used registration algorithms that need to match image histogram statistics to the more densely labeled histological brain atlases. The standard method is a multiscale Mutual Information B-spline algorithm that dynamically generates an average template as an intermediate registration target. We determined that this method performs poorly when registering CLARITY brains to the Allen Institute's Mouse Reference Atlas (ARA), because the image histogram statistics are poorly matched. Therefore, we developed a method (Mask-LDDMM) for registering CLARITY images, that automatically find the brain boundary and learns the optimal deformation between the brain and atlas masks. Using Mask-LDDMM without an average template provided better results than the standard approach when registering CLARITY brains to the ARA. The LDDMM pipelines developed here provide a fast automated way to anatomically annotate CLARITY images. Our code is available as open source software at http://NeuroData.io.
VESICLE: Volumetric Evaluation of Synaptic Interfaces using Computer vision at Large Scale
Sep 07, 2015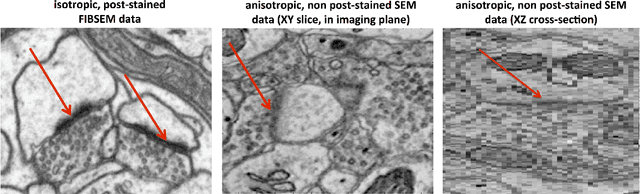

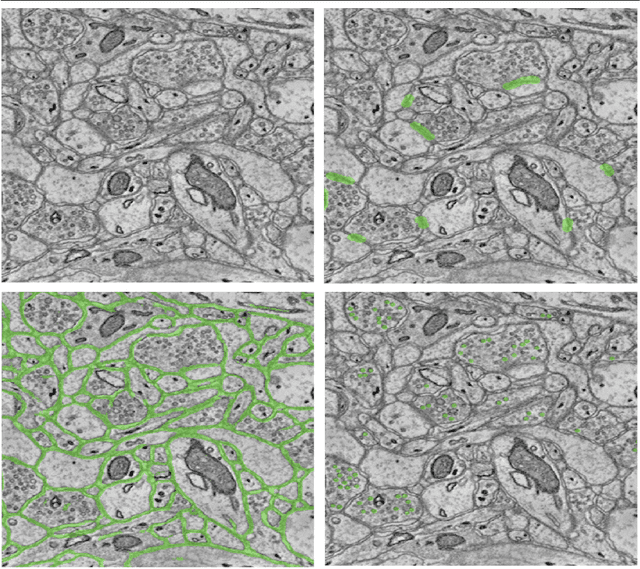
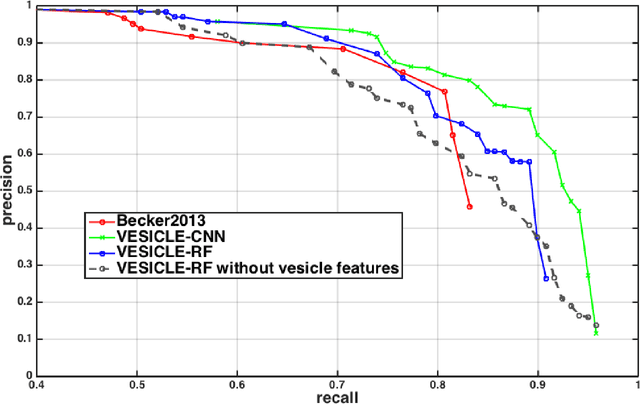
An open challenge problem at the forefront of modern neuroscience is to obtain a comprehensive mapping of the neural pathways that underlie human brain function; an enhanced understanding of the wiring diagram of the brain promises to lead to new breakthroughs in diagnosing and treating neurological disorders. Inferring brain structure from image data, such as that obtained via electron microscopy (EM), entails solving the problem of identifying biological structures in large data volumes. Synapses, which are a key communication structure in the brain, are particularly difficult to detect due to their small size and limited contrast. Prior work in automated synapse detection has relied upon time-intensive biological preparations (post-staining, isotropic slice thicknesses) in order to simplify the problem. This paper presents VESICLE, the first known approach designed for mammalian synapse detection in anisotropic, non-post-stained data. Our methods explicitly leverage biological context, and the results exceed existing synapse detection methods in terms of accuracy and scalability. We provide two different approaches - one a deep learning classifier (VESICLE-CNN) and one a lightweight Random Forest approach (VESICLE-RF) to offer alternatives in the performance-scalability space. Addressing this synapse detection challenge enables the analysis of high-throughput imaging data soon expected to reach petabytes of data, and provide tools for more rapid estimation of brain-graphs. Finally, to facilitate community efforts, we developed tools for large-scale object detection, and demonstrated this framework to find $\approx$ 50,000 synapses in 60,000 $\mu m ^3$ (220 GB on disk) of electron microscopy data.
* v4: added clarifying figures and updates for readability. v3: fixed metadata. 11 pp v2: Added CNN classifier, significant changes to improve performance and generalization
An Automated Images-to-Graphs Framework for High Resolution Connectomics
Apr 30, 2015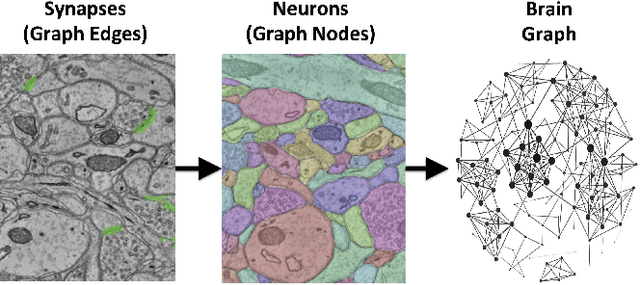
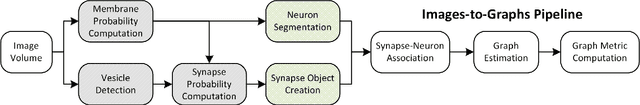
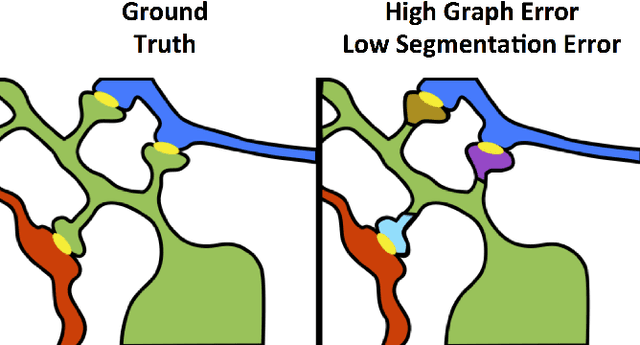
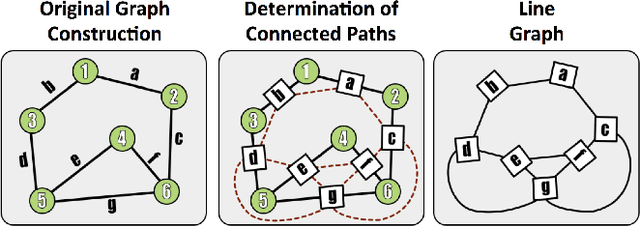
Reconstructing a map of neuronal connectivity is a critical challenge in contemporary neuroscience. Recent advances in high-throughput serial section electron microscopy (EM) have produced massive 3D image volumes of nanoscale brain tissue for the first time. The resolution of EM allows for individual neurons and their synaptic connections to be directly observed. Recovering neuronal networks by manually tracing each neuronal process at this scale is unmanageable, and therefore researchers are developing automated image processing modules. Thus far, state-of-the-art algorithms focus only on the solution to a particular task (e.g., neuron segmentation or synapse identification). In this manuscript we present the first fully automated images-to-graphs pipeline (i.e., a pipeline that begins with an imaged volume of neural tissue and produces a brain graph without any human interaction). To evaluate overall performance and select the best parameters and methods, we also develop a metric to assess the quality of the output graphs. We evaluate a set of algorithms and parameters, searching possible operating points to identify the best available brain graph for our assessment metric. Finally, we deploy a reference end-to-end version of the pipeline on a large, publicly available data set. This provides a baseline result and framework for community analysis and future algorithm development and testing. All code and data derivatives have been made publicly available toward eventually unlocking new biofidelic computational primitives and understanding of neuropathologies.
Spectral Clustering for Divide-and-Conquer Graph Matching
Mar 12, 2015We present a parallelized bijective graph matching algorithm that leverages seeds and is designed to match very large graphs. Our algorithm combines spectral graph embedding with existing state-of-the-art seeded graph matching procedures. We justify our approach by proving that modestly correlated, large stochastic block model random graphs are correctly matched utilizing very few seeds through our divide-and-conquer procedure. We also demonstrate the effectiveness of our approach in matching very large graphs in simulated and real data examples, showing up to a factor of 8 improvement in runtime with minimal sacrifice in accuracy.