 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSpeech enhancement deep-learning architecture for efficient edge processing
May 27, 2024


Deep learning has become a de facto method of choice for speech enhancement tasks with significant improvements in speech quality. However, real-time processing with reduced size and computations for low-power edge devices drastically degrades speech quality. Recently, transformer-based architectures have greatly reduced the memory requirements and provided ways to improve the model performance through local and global contexts. However, the transformer operations remain computationally heavy. In this work, we introduce WaveUNet squeeze-excitation Res2 (WSR)-based metric generative adversarial network (WSR-MGAN) architecture that can be efficiently implemented on low-power edge devices for noise suppression tasks while maintaining speech quality. We utilize multi-scale features using Res2Net blocks that can be related to spectral content used in speech-processing tasks. In the generator, we integrate squeeze-excitation blocks (SEB) with multi-scale features for maintaining local and global contexts along with gated recurrent units (GRUs). The proposed approach is optimized through a combined loss function calculated over raw waveform, multi-resolution magnitude spectrogram, and objective metrics using a metric discriminator. Experimental results in terms of various objective metrics on VoiceBank+DEMAND and DNS-2020 challenge datasets demonstrate that the proposed speech enhancement (SE) approach outperforms the baselines and achieves state-of-the-art (SOTA) performance in the time domain.
Equivariant Graph Neural Operator for Modeling 3D Dynamics
Jan 19, 2024



Modeling the complex three-dimensional (3D) dynamics of relational systems is an important problem in the natural sciences, with applications ranging from molecular simulations to particle mechanics. Machine learning methods have achieved good success by learning graph neural networks to model spatial interactions. However, these approaches do not faithfully capture temporal correlations since they only model next-step predictions. In this work, we propose Equivariant Graph Neural Operator (EGNO), a novel and principled method that directly models dynamics as trajectories instead of just next-step prediction. Different from existing methods, EGNO explicitly learns the temporal evolution of 3D dynamics where we formulate the dynamics as a function over time and learn neural operators to approximate it. To capture the temporal correlations while keeping the intrinsic SE(3)-equivariance, we develop equivariant temporal convolutions parameterized in the Fourier space and build EGNO by stacking the Fourier layers over equivariant networks. EGNO is the first operator learning framework that is capable of modeling solution dynamics functions over time while retaining 3D equivariance. Comprehensive experiments in multiple domains, including particle simulations, human motion capture, and molecular dynamics, demonstrate the significantly superior performance of EGNO against existing methods, thanks to the equivariant temporal modeling.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
Linking the Dynamic PicoProbe Analytical Electron-Optical Beam Line / Microscope to Supercomputers
Aug 25, 2023



The Dynamic PicoProbe at Argonne National Laboratory is undergoing upgrades that will enable it to produce up to 100s of GB of data per day. While this data is highly important for both fundamental science and industrial applications, there is currently limited on-site infrastructure to handle these high-volume data streams. We address this problem by providing a software architecture capable of supporting large-scale data transfers to the neighboring supercomputers at the Argonne Leadership Computing Facility. To prepare for future scientific workflows, we implement two instructive use cases for hyperspectral and spatiotemporal datasets, which include: (i) off-site data transfer, (ii) machine learning/artificial intelligence and traditional data analysis approaches, and (iii) automatic metadata extraction and cataloging of experimental results. This infrastructure supports expected workloads and also provides domain scientists the ability to reinterrogate data from past experiments to yield additional scientific value and derive new insights.
Towards a Modular Architecture for Science Factories
Aug 18, 2023Advances in robotic automation, high-performance computing (HPC), and artificial intelligence (AI) encourage us to conceive of science factories: large, general-purpose computation- and AI-enabled self-driving laboratories (SDLs) with the generality and scale needed both to tackle large discovery problems and to support thousands of scientists. Science factories require modular hardware and software that can be replicated for scale and (re)configured to support many applications. To this end, we propose a prototype modular science factory architecture in which reconfigurable modules encapsulating scientific instruments are linked with manipulators to form workcells, that can themselves be combined to form larger assemblages, and linked with distributed computing for simulation, AI model training and inference, and related tasks. Workflows that perform sets of actions on modules can be specified, and various applications, comprising workflows plus associated computational and data manipulation steps, can be run concurrently. We report on our experiences prototyping this architecture and applying it in experiments involving 15 different robotic apparatus, five applications (one in education, two in biology, two in materials), and a variety of workflows, across four laboratories. We describe the reuse of modules, workcells, and workflows in different applications, the migration of applications between workcells, and the use of digital twins, and suggest directions for future work aimed at yet more generality and scalability. Code and data are available at https://ad-sdl.github.io/wei2023 and in the Supplementary Information
Transferable Graph Neural Fingerprint Models for Quick Response to Future Bio-Threats
Jul 17, 2023



Fast screening of drug molecules based on the ligand binding affinity is an important step in the drug discovery pipeline. Graph neural fingerprint is a promising method for developing molecular docking surrogates with high throughput and great fidelity. In this study, we built a COVID-19 drug docking dataset of about 300,000 drug candidates on 23 coronavirus protein targets. With this dataset, we trained graph neural fingerprint docking models for high-throughput virtual COVID-19 drug screening. The graph neural fingerprint models yield high prediction accuracy on docking scores with the mean squared error lower than $0.21$ kcal/mol for most of the docking targets, showing significant improvement over conventional circular fingerprint methods. To make the neural fingerprints transferable for unknown targets, we also propose a transferable graph neural fingerprint method trained on multiple targets. With comparable accuracy to target-specific graph neural fingerprint models, the transferable model exhibits superb training and data efficiency. We highlight that the impact of this study extends beyond COVID-19 dataset, as our approach for fast virtual ligand screening can be easily adapted and integrated into a general machine learning-accelerated pipeline to battle future bio-threats.
On the Robustness of AlphaFold: A COVID-19 Case Study
Jan 12, 2023



Protein folding neural networks (PFNNs) such as AlphaFold predict remarkably accurate structures of proteins compared to other approaches. However, the robustness of such networks has heretofore not been explored. This is particularly relevant given the broad social implications of such technologies and the fact that biologically small perturbations in the protein sequence do not generally lead to drastic changes in the protein structure. In this paper, we demonstrate that AlphaFold does not exhibit such robustness despite its high accuracy. This raises the challenge of detecting and quantifying the extent to which these predicted protein structures can be trusted. To measure the robustness of the predicted structures, we utilize (i) the root-mean-square deviation (RMSD) and (ii) the Global Distance Test (GDT) similarity measure between the predicted structure of the original sequence and the structure of its adversarially perturbed version. We prove that the problem of minimally perturbing protein sequences to fool protein folding neural networks is NP-complete. Based on the well-established BLOSUM62 sequence alignment scoring matrix, we generate adversarial protein sequences and show that the RMSD between the predicted protein structure and the structure of the original sequence are very large when the adversarial changes are bounded by (i) 20 units in the BLOSUM62 distance, and (ii) five residues (out of hundreds or thousands of residues) in the given protein sequence. In our experimental evaluation, we consider 111 COVID-19 proteins in the Universal Protein resource (UniProt), a central resource for protein data managed by the European Bioinformatics Institute, Swiss Institute of Bioinformatics, and the US Protein Information Resource. These result in an overall GDT similarity test score average of around 34%, demonstrating a substantial drop in the performance of AlphaFold.
Deep Surrogate Docking: Accelerating Automated Drug Discovery with Graph Neural Networks
Nov 04, 2022



The process of screening molecules for desirable properties is a key step in several applications, ranging from drug discovery to material design. During the process of drug discovery specifically, protein-ligand docking, or chemical docking, is a standard in-silico scoring technique that estimates the binding affinity of molecules with a specific protein target. Recently, however, as the number of virtual molecules available to test has rapidly grown, these classical docking algorithms have created a significant computational bottleneck. We address this problem by introducing Deep Surrogate Docking (DSD), a framework that applies deep learning-based surrogate modeling to accelerate the docking process substantially. DSD can be interpreted as a formalism of several earlier surrogate prefiltering techniques, adding novel metrics and practical training practices. Specifically, we show that graph neural networks (GNNs) can serve as fast and accurate estimators of classical docking algorithms. Additionally, we introduce FiLMv2, a novel GNN architecture which we show outperforms existing state-of-the-art GNN architectures, attaining more accurate and stable performance by allowing the model to filter out irrelevant information from data more efficiently. Through extensive experimentation and analysis, we show that the DSD workflow combined with the FiLMv2 architecture provides a 9.496x speedup in molecule screening with a <3% recall error rate on an example docking task. Our open-source code is available at https://github.com/ryienh/graph-dock.
Protein Folding Neural Networks Are Not Robust
Sep 19, 2021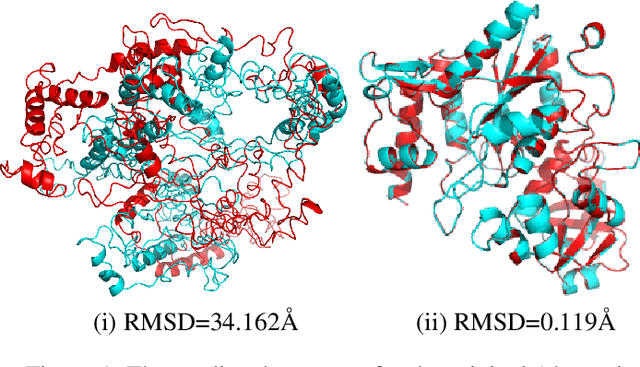
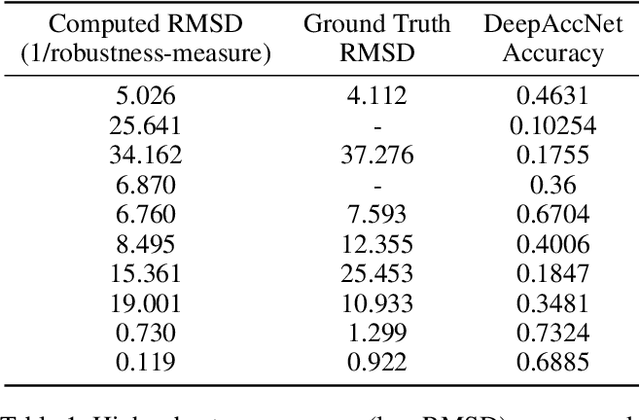
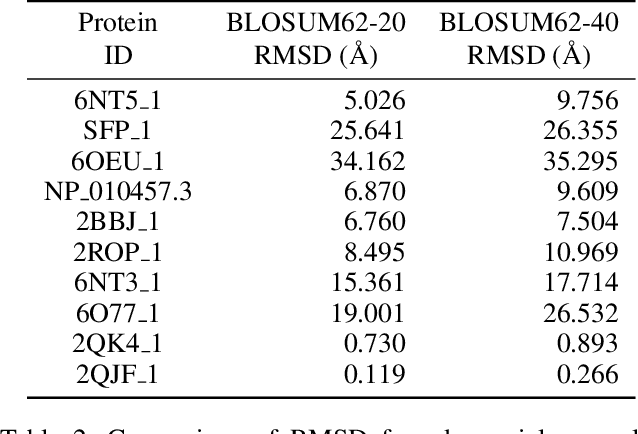
Deep neural networks such as AlphaFold and RoseTTAFold predict remarkably accurate structures of proteins compared to other algorithmic approaches. It is known that biologically small perturbations in the protein sequence do not lead to drastic changes in the protein structure. In this paper, we demonstrate that RoseTTAFold does not exhibit such a robustness despite its high accuracy, and biologically small perturbations for some input sequences result in radically different predicted protein structures. This raises the challenge of detecting when these predicted protein structures cannot be trusted. We define the robustness measure for the predicted structure of a protein sequence to be the inverse of the root-mean-square distance (RMSD) in the predicted structure and the structure of its adversarially perturbed sequence. We use adversarial attack methods to create adversarial protein sequences, and show that the RMSD in the predicted protein structure ranges from 0.119\r{A} to 34.162\r{A} when the adversarial perturbations are bounded by 20 units in the BLOSUM62 distance. This demonstrates very high variance in the robustness measure of the predicted structures. We show that the magnitude of the correlation (0.917) between our robustness measure and the RMSD between the predicted structure and the ground truth is high, that is, the predictions with low robustness measure cannot be trusted. This is the first paper demonstrating the susceptibility of RoseTTAFold to adversarial attacks.
CrossedWires: A Dataset of Syntactically Equivalent but Semantically Disparate Deep Learning Models
Aug 29, 2021

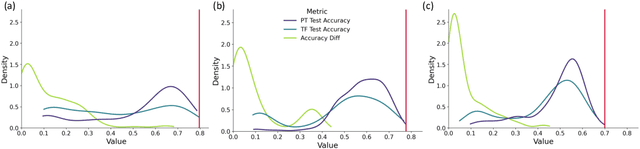

The training of neural networks using different deep learning frameworks may lead to drastically differing accuracy levels despite the use of the same neural network architecture and identical training hyperparameters such as learning rate and choice of optimization algorithms. Currently, our ability to build standardized deep learning models is limited by the availability of a suite of neural network and corresponding training hyperparameter benchmarks that expose differences between existing deep learning frameworks. In this paper, we present a living dataset of models and hyperparameters, called CrossedWires, that exposes semantic differences between two popular deep learning frameworks: PyTorch and Tensorflow. The CrossedWires dataset currently consists of models trained on CIFAR10 images using three different computer vision architectures: VGG16, ResNet50 and DenseNet121 across a large hyperparameter space. Using hyperparameter optimization, each of the three models was trained on 400 sets of hyperparameters suggested by the HyperSpace search algorithm. The CrossedWires dataset includes PyTorch and Tensforflow models with test accuracies as different as 0.681 on syntactically equivalent models and identical hyperparameter choices. The 340 GB dataset and benchmarks presented here include the performance statistics, training curves, and model weights for all 1200 hyperparameter choices, resulting in 2400 total models. The CrossedWires dataset provides an opportunity to study semantic differences between syntactically equivalent models across popular deep learning frameworks. Further, the insights obtained from this study can enable the development of algorithms and tools that improve reliability and reproducibility of deep learning frameworks. The dataset is freely available at https://github.com/maxzvyagin/crossedwires through a Python API and direct download link.