 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
Transferable Graph Neural Fingerprint Models for Quick Response to Future Bio-Threats
Jul 17, 2023



Fast screening of drug molecules based on the ligand binding affinity is an important step in the drug discovery pipeline. Graph neural fingerprint is a promising method for developing molecular docking surrogates with high throughput and great fidelity. In this study, we built a COVID-19 drug docking dataset of about 300,000 drug candidates on 23 coronavirus protein targets. With this dataset, we trained graph neural fingerprint docking models for high-throughput virtual COVID-19 drug screening. The graph neural fingerprint models yield high prediction accuracy on docking scores with the mean squared error lower than $0.21$ kcal/mol for most of the docking targets, showing significant improvement over conventional circular fingerprint methods. To make the neural fingerprints transferable for unknown targets, we also propose a transferable graph neural fingerprint method trained on multiple targets. With comparable accuracy to target-specific graph neural fingerprint models, the transferable model exhibits superb training and data efficiency. We highlight that the impact of this study extends beyond COVID-19 dataset, as our approach for fast virtual ligand screening can be easily adapted and integrated into a general machine learning-accelerated pipeline to battle future bio-threats.
Deep learning methods for drug response prediction in cancer: predominant and emerging trends
Nov 18, 2022Cancer claims millions of lives yearly worldwide. While many therapies have been made available in recent years, by in large cancer remains unsolved. Exploiting computational predictive models to study and treat cancer holds great promise in improving drug development and personalized design of treatment plans, ultimately suppressing tumors, alleviating suffering, and prolonging lives of patients. A wave of recent papers demonstrates promising results in predicting cancer response to drug treatments while utilizing deep learning methods. These papers investigate diverse data representations, neural network architectures, learning methodologies, and evaluations schemes. However, deciphering promising predominant and emerging trends is difficult due to the variety of explored methods and lack of standardized framework for comparing drug response prediction models. To obtain a comprehensive landscape of deep learning methods, we conducted an extensive search and analysis of deep learning models that predict the response to single drug treatments. A total of 60 deep learning-based models have been curated and summary plots were generated. Based on the analysis, observable patterns and prevalence of methods have been revealed. This review allows to better understand the current state of the field and identify major challenges and promising solution paths.
Deep Surrogate Docking: Accelerating Automated Drug Discovery with Graph Neural Networks
Nov 04, 2022



The process of screening molecules for desirable properties is a key step in several applications, ranging from drug discovery to material design. During the process of drug discovery specifically, protein-ligand docking, or chemical docking, is a standard in-silico scoring technique that estimates the binding affinity of molecules with a specific protein target. Recently, however, as the number of virtual molecules available to test has rapidly grown, these classical docking algorithms have created a significant computational bottleneck. We address this problem by introducing Deep Surrogate Docking (DSD), a framework that applies deep learning-based surrogate modeling to accelerate the docking process substantially. DSD can be interpreted as a formalism of several earlier surrogate prefiltering techniques, adding novel metrics and practical training practices. Specifically, we show that graph neural networks (GNNs) can serve as fast and accurate estimators of classical docking algorithms. Additionally, we introduce FiLMv2, a novel GNN architecture which we show outperforms existing state-of-the-art GNN architectures, attaining more accurate and stable performance by allowing the model to filter out irrelevant information from data more efficiently. Through extensive experimentation and analysis, we show that the DSD workflow combined with the FiLMv2 architecture provides a 9.496x speedup in molecule screening with a <3% recall error rate on an example docking task. Our open-source code is available at https://github.com/ryienh/graph-dock.
Protein-Ligand Docking Surrogate Models: A SARS-CoV-2 Benchmark for Deep Learning Accelerated Virtual Screening
Jun 30, 2021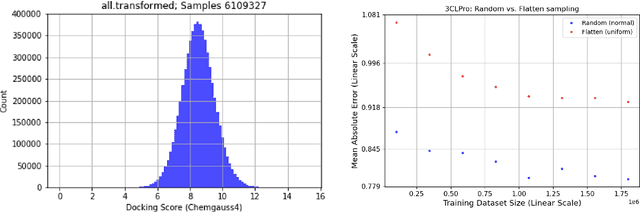

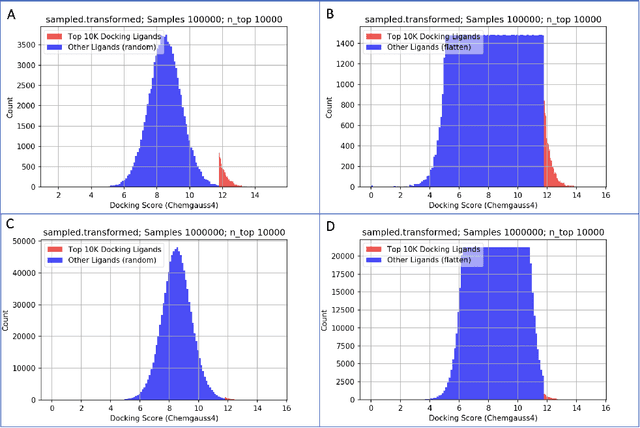
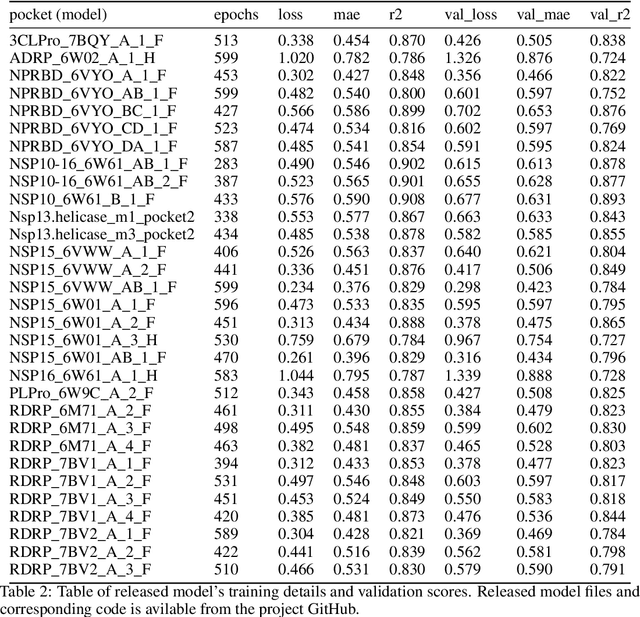
We propose a benchmark to study surrogate model accuracy for protein-ligand docking. We share a dataset consisting of 200 million 3D complex structures and 2D structure scores across a consistent set of 13 million "in-stock" molecules over 15 receptors, or binding sites, across the SARS-CoV-2 proteome. Our work shows surrogate docking models have six orders of magnitude more throughput than standard docking protocols on the same supercomputer node types. We demonstrate the power of high-speed surrogate models by running each target against 1 billion molecules in under a day (50k predictions per GPU seconds). We showcase a workflow for docking utilizing surrogate ML models as a pre-filter. Our workflow is ten times faster at screening a library of compounds than the standard technique, with an error rate less than 0.01\% of detecting the underlying best scoring 0.1\% of compounds. Our analysis of the speedup explains that to screen more molecules under a docking paradigm, another order of magnitude speedup must come from model accuracy rather than computing speed (which, if increased, will not anymore alter our throughput to screen molecules). We believe this is strong evidence for the community to begin focusing on improving the accuracy of surrogate models to improve the ability to screen massive compound libraries 100x or even 1000x faster than current techniques.
Spatial Graph Attention and Curiosity-driven Policy for Antiviral Drug Discovery
Jun 20, 2021
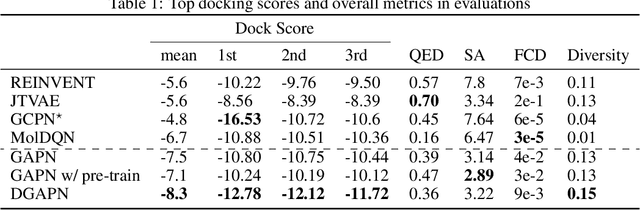
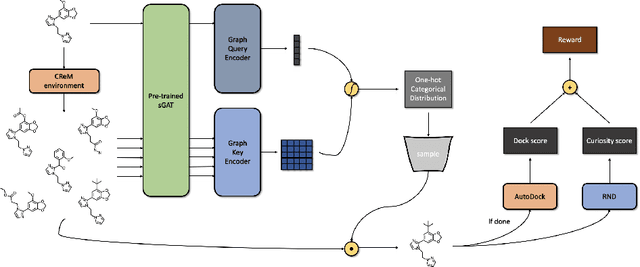
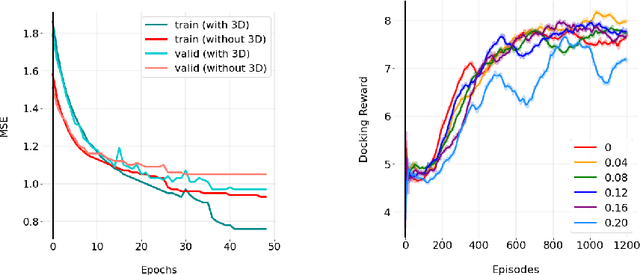
We developed Distilled Graph Attention Policy Networks (DGAPNs), a curiosity-driven reinforcement learning model to generate novel graph-structured chemical representations that optimize user-defined objectives by efficiently navigating a physically constrained domain. The framework is examined on the task of generating molecules that are designed to bind, noncovalently, to functional sites of SARS-CoV-2 proteins. We present a spatial Graph Attention Network (sGAT) that leverages self-attention over both node and edge attributes as well as encoding spatial structure -- this capability is of considerable interest in areas such as molecular and synthetic biology and drug discovery. An attentional policy network is then introduced to learn decision rules for a dynamic, fragment-based chemical environment, and state-of-the-art policy gradient techniques are employed to train the network with enhanced stability. Exploration is efficiently encouraged by incorporating innovation reward bonuses learned and proposed by random network distillation. In experiments, our framework achieved outstanding results compared to state-of-the-art algorithms, while increasing the diversity of proposed molecules and reducing the complexity of paths to chemical synthesis.
Scaffold Embeddings: Learning the Structure Spanned by Chemical Fragments, Scaffolds and Compounds
Mar 11, 2021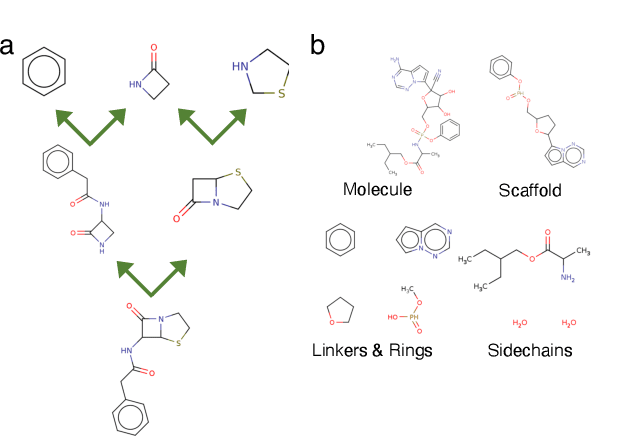

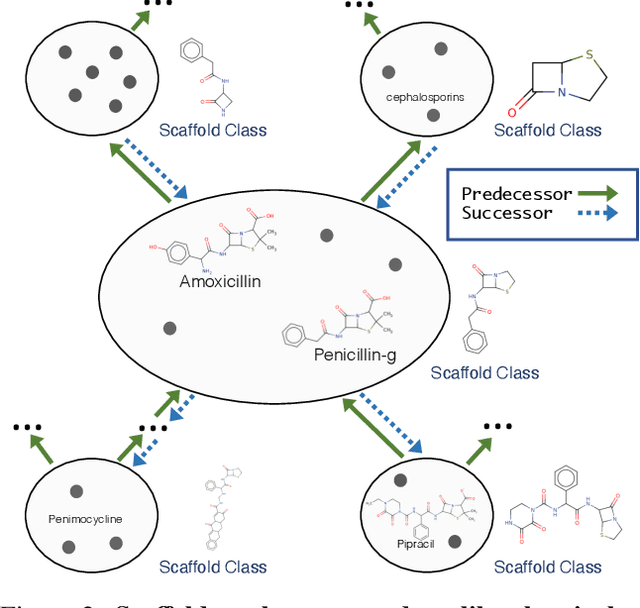
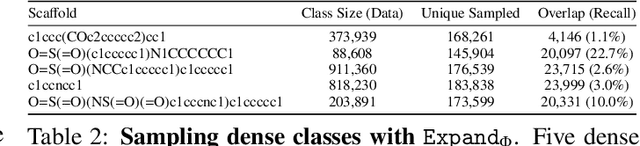
Molecules have seemed like a natural fit to deep learning's tendency to handle a complex structure through representation learning, given enough data. However, this often continuous representation is not natural for understanding chemical space as a domain and is particular to samples and their differences. We focus on exploring a natural structure for representing chemical space as a structured domain: embedding drug-like chemical space into an enumerable hypergraph based on scaffold classes linked through an inclusion operator. This paper shows how molecules form classes of scaffolds, how scaffolds relate to each in a hypergraph, and how this structure of scaffolds is natural for drug discovery workflows such as predicting properties and optimizing molecular structures. We compare the assumptions and utility of various embeddings of molecules, such as their respective induced distance metrics, their extendibility to represent chemical space as a structured domain, and the consequences of utilizing the structure for learning tasks.
Learning Curves for Drug Response Prediction in Cancer Cell Lines
Nov 25, 2020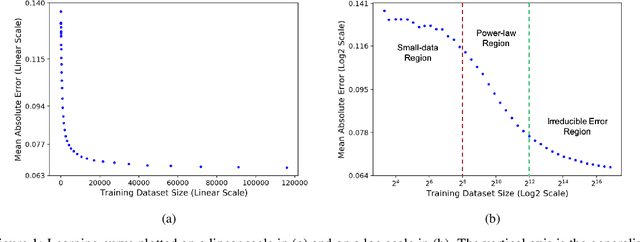
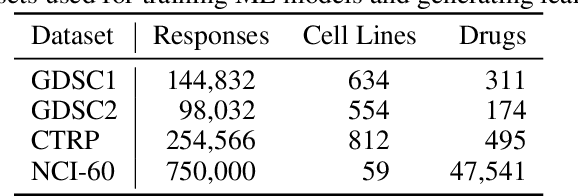
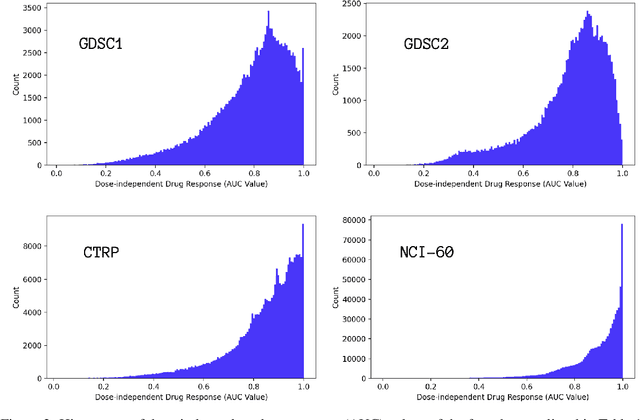
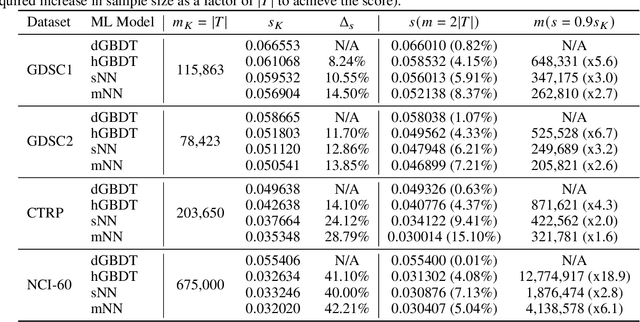
Motivated by the size of cell line drug sensitivity data, researchers have been developing machine learning (ML) models for predicting drug response to advance cancer treatment. As drug sensitivity studies continue generating data, a common question is whether the proposed predictors can further improve the generalization performance with more training data. We utilize empirical learning curves for evaluating and comparing the data scaling properties of two neural networks (NNs) and two gradient boosting decision tree (GBDT) models trained on four drug screening datasets. The learning curves are accurately fitted to a power law model, providing a framework for assessing the data scaling behavior of these predictors. The curves demonstrate that no single model dominates in terms of prediction performance across all datasets and training sizes, suggesting that the shape of these curves depends on the unique model-dataset pair. The multi-input NN (mNN), in which gene expressions and molecular drug descriptors are input into separate subnetworks, outperforms a single-input NN (sNN), where the cell and drug features are concatenated for the input layer. In contrast, a GBDT with hyperparameter tuning exhibits superior performance as compared with both NNs at the lower range of training sizes for two of the datasets, whereas the mNN performs better at the higher range of training sizes. Moreover, the trajectory of the curves suggests that increasing the sample size is expected to further improve prediction scores of both NNs. These observations demonstrate the benefit of using learning curves to evaluate predictors, providing a broader perspective on the overall data scaling characteristics. The fitted power law curves provide a forward-looking performance metric and can serve as a co-design tool to guide experimental biologists and computational scientists in the design of future experiments.
Regression Enrichment Surfaces: a Simple Analysis Technique for Virtual Drug Screening Models
Jun 01, 2020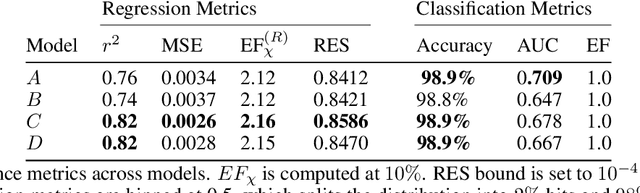
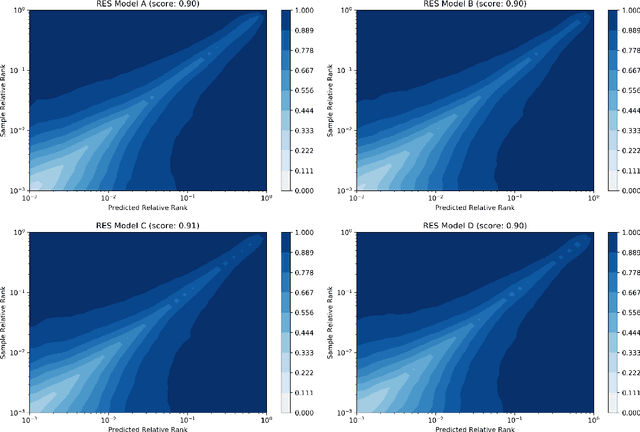
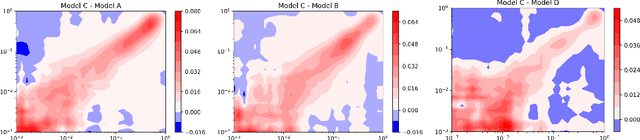
We present a new method for understanding the performance of a model in virtual drug screening tasks. While most virtual screening problems present as a mix between ranking and classification, the models are typically trained as regression models presenting a problem requiring either a choice of a cutoff or ranking measure. Our method, regression enrichment surfaces (RES), is based on the goal of virtual screening: to detect as many of the top-performing treatments as possible. We outline history of virtual screening performance measures and the idea behind RES. We offer a python package and details on how to implement and interpret the results.
Targeting SARS-CoV-2 with AI- and HPC-enabled Lead Generation: A First Data Release
May 28, 2020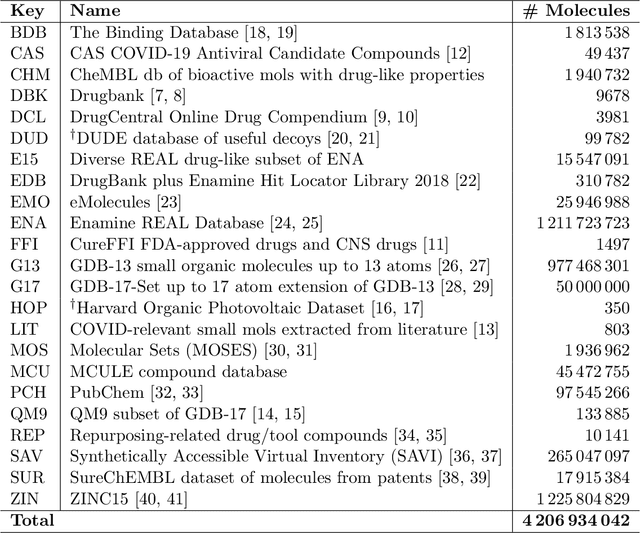
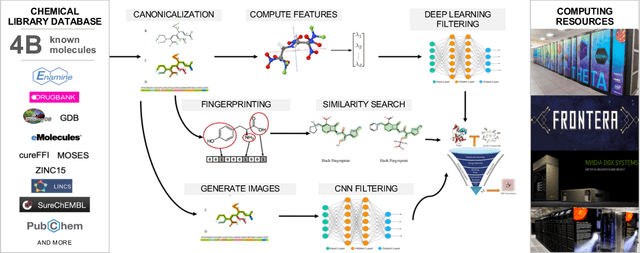

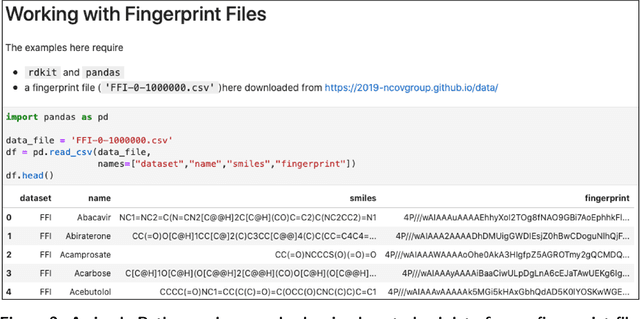
Researchers across the globe are seeking to rapidly repurpose existing drugs or discover new drugs to counter the the novel coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). One promising approach is to train machine learning (ML) and artificial intelligence (AI) tools to screen large numbers of small molecules. As a contribution to that effort, we are aggregating numerous small molecules from a variety of sources, using high-performance computing (HPC) to computer diverse properties of those molecules, using the computed properties to train ML/AI models, and then using the resulting models for screening. In this first data release, we make available 23 datasets collected from community sources representing over 4.2 B molecules enriched with pre-computed: 1) molecular fingerprints to aid similarity searches, 2) 2D images of molecules to enable exploration and application of image-based deep learning methods, and 3) 2D and 3D molecular descriptors to speed development of machine learning models. This data release encompasses structural information on the 4.2 B molecules and 60 TB of pre-computed data. Future releases will expand the data to include more detailed molecular simulations, computed models, and other products.