 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtDBench: A Unified Benchmark of Protein Binder Design and Evaluation
May 05, 2026Recent advances in de novo protein binder design have enabled increasing experimental validation, yet reported in silico metrics remain difficult to interpret or compare across studies due to non-standardized evaluation protocols. We introduce ProtDBench, a standardized and throughput-aware evaluation framework for protein binder design. ProtDBench defines unified benchmark tasks, evaluation protocols, and success criteria, enabling systematic analysis of how evaluation design influences observed performance. Using a large wet-lab annotated dataset, we analyze commonly used structure prediction models as evaluation verifiers, revealing substantial verifier-dependent bias and limited agreement under identical filtering protocols. We then benchmark representative open-source generative binder design methods across ten diverse protein targets under a fixed evaluation protocol. Beyond per-sequence success rates, ProtDBench incorporates throughput-aware metrics based on a fixed 24-hour budget, as well as cluster-level success criteria to account for structural diversity. Together, these results expose systematic differences induced by filtering rules, success definitions, and throughput-aware evaluation between computational efficiency, success rate, and structural diversity. Overall, ProtDBench provides a fair and reproducible evaluation pipeline that supports systematic and controlled comparison of protein binder design methods under realistic evaluation settings.
A-CODE: Fully Atomic Protein Co-Design with Unified Multimodal Diffusion
May 05, 2026We present A-CODE, a fully atomic unified one-stage protein co-design model that simultaneously refines discrete atom types and continuous atom coordinates. Unlike predominant two-stage methods that cascade structure design with amino acid-level sequence design, our approach is fully atomic within a unified multimodal diffusion framework, in which residue identities are inferred solely from atom-level predictions. Built upon the powerful all-atom architecture, A-CODE achieves superior designability for unconditional protein generation, outperforming all existing one-stage and two-stage design models. For binder design, A-CODE rivals and even outperforms existing state-of-the-art two-stage design models and, compared with the existing one-stage co-design model, achieves a drastic tenfold improvement in success rate on hard tasks. The inherent flexibility of our atomic formulation enables, for the first time, seamless adaptation to non-canonical amino acid (ncAA) modeling. Our fully atomic framework establishes a new, versatile foundation for all-atom generative modeling that can be naturally extended to complex biomolecular systems.
Protenix-Mini: Efficient Structure Predictor via Compact Architecture, Few-Step Diffusion and Switchable pLM
Jul 16, 2025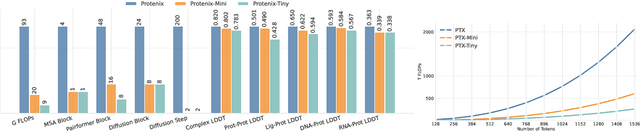

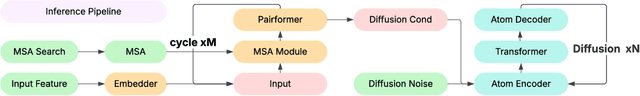

Lightweight inference is critical for biomolecular structure prediction and other downstream tasks, enabling efficient real-world deployment and inference-time scaling for large-scale applications. In this work, we address the challenge of balancing model efficiency and prediction accuracy by making several key modifications, 1) Multi-step AF3 sampler is replaced by a few-step ODE sampler, significantly reducing computational overhead for the diffusion module part during inference; 2) In the open-source Protenix framework, a subset of pairformer or diffusion transformer blocks doesn't make contributions to the final structure prediction, presenting opportunities for architectural pruning and lightweight redesign; 3) A model incorporating an ESM module is trained to substitute the conventional MSA module, reducing MSA preprocessing time. Building on these key insights, we present Protenix-Mini, a compact and optimized model designed for efficient protein structure prediction. This streamlined version incorporates a more efficient architectural design with a two-step Ordinary Differential Equation (ODE) sampling strategy. By eliminating redundant Transformer components and refining the sampling process, Protenix-Mini significantly reduces model complexity with slight accuracy drop. Evaluations on benchmark datasets demonstrate that it achieves high-fidelity predictions, with only a negligible 1 to 5 percent decrease in performance on benchmark datasets compared to its full-scale counterpart. This makes Protenix-Mini an ideal choice for applications where computational resources are limited but accurate structure prediction remains crucial.
Graph Condensation via Receptive Field Distribution Matching
Jun 28, 2022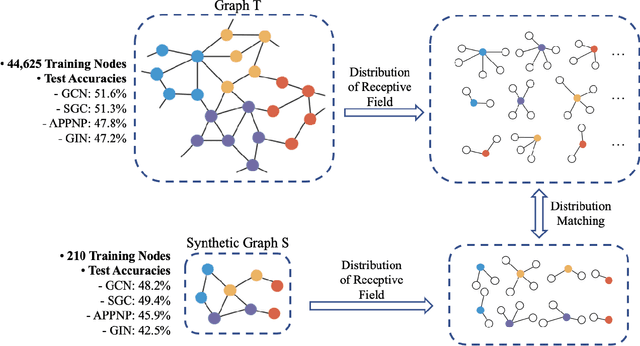


Graph neural networks (GNNs) enable the analysis of graphs using deep learning, with promising results in capturing structured information in graphs. This paper focuses on creating a small graph to represent the original graph, so that GNNs trained on the size-reduced graph can make accurate predictions. We view the original graph as a distribution of receptive fields and aim to synthesize a small graph whose receptive fields share a similar distribution. Thus, we propose Graph Condesation via Receptive Field Distribution Matching (GCDM), which is accomplished by optimizing the synthetic graph through the use of a distribution matching loss quantified by maximum mean discrepancy (MMD). Additionally, we demonstrate that the synthetic graph generated by GCDM is highly generalizable to a variety of models in evaluation phase and that the condensing speed is significantly improved using this framework.
uGLAD: Sparse graph recovery by optimizing deep unrolled networks
May 23, 2022
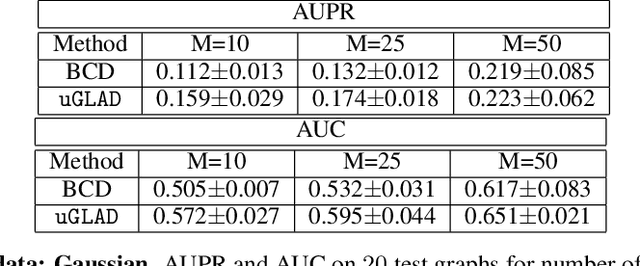
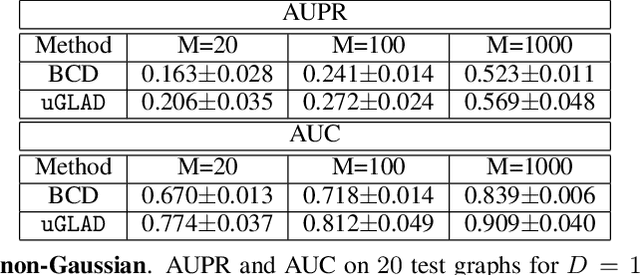

Probabilistic Graphical Models (PGMs) are generative models of complex systems. They rely on conditional independence assumptions between variables to learn sparse representations which can be visualized in a form of a graph. Such models are used for domain exploration and structure discovery in poorly understood domains. This work introduces a novel technique to perform sparse graph recovery by optimizing deep unrolled networks. Assuming that the input data $X\in\mathbb{R}^{M\times D}$ comes from an underlying multivariate Gaussian distribution, we apply a deep model on $X$ that outputs the precision matrix $\Theta$, which can also be interpreted as the adjacency matrix. Our model, uGLAD, builds upon and extends the state-of-the-art model GLAD to the unsupervised setting. The key benefits of our model are (1) uGLAD automatically optimizes sparsity-related regularization parameters leading to better performance than existing algorithms. (2) We introduce multi-task learning based `consensus' strategy for robust handling of missing data in an unsupervised setting. We evaluate model results on synthetic Gaussian data, non-Gaussian data generated from Gene Regulatory Networks, and present a case study in anaerobic digestion.
Efficient Dynamic Graph Representation Learning at Scale
Dec 14, 2021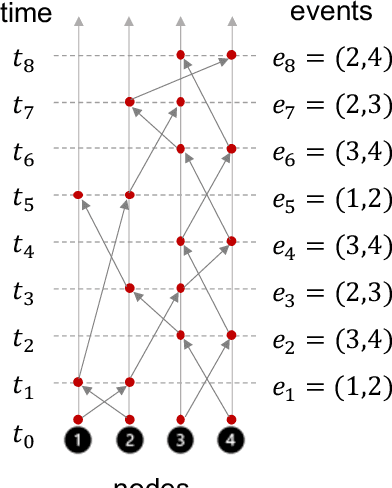
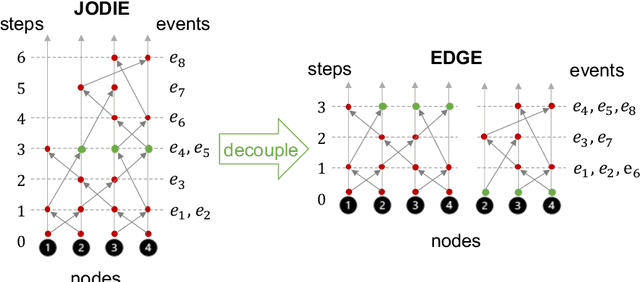
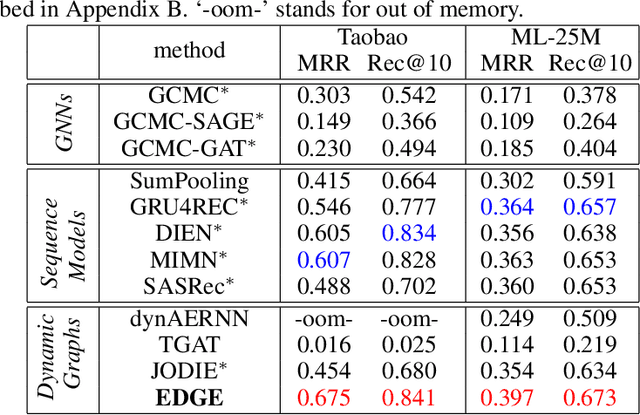
Dynamic graphs with ordered sequences of events between nodes are prevalent in real-world industrial applications such as e-commerce and social platforms. However, representation learning for dynamic graphs has posed great computational challenges due to the time and structure dependency and irregular nature of the data, preventing such models from being deployed to real-world applications. To tackle this challenge, we propose an efficient algorithm, Efficient Dynamic Graph lEarning (EDGE), which selectively expresses certain temporal dependency via training loss to improve the parallelism in computations. We show that EDGE can scale to dynamic graphs with millions of nodes and hundreds of millions of temporal events and achieve new state-of-the-art (SOTA) performance.
Multi-task Learning of Order-Consistent Causal Graphs
Nov 03, 2021


We consider the problem of discovering $K$ related Gaussian directed acyclic graphs (DAGs), where the involved graph structures share a consistent causal order and sparse unions of supports. Under the multi-task learning setting, we propose a $l_1/l_2$-regularized maximum likelihood estimator (MLE) for learning $K$ linear structural equation models. We theoretically show that the joint estimator, by leveraging data across related tasks, can achieve a better sample complexity for recovering the causal order (or topological order) than separate estimations. Moreover, the joint estimator is able to recover non-identifiable DAGs, by estimating them together with some identifiable DAGs. Lastly, our analysis also shows the consistency of union support recovery of the structures. To allow practical implementation, we design a continuous optimization problem whose optimizer is the same as the joint estimator and can be approximated efficiently by an iterative algorithm. We validate the theoretical analysis and the effectiveness of the joint estimator in experiments.
Understanding Deep Architectures with Reasoning Layer
Jun 24, 2020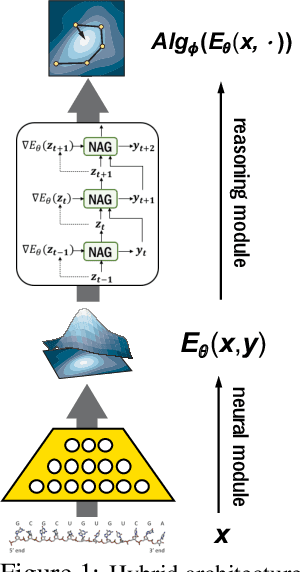

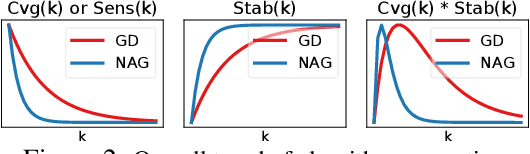
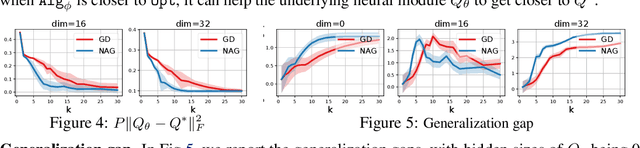
Recently, there has been a surge of interest in combining deep learning models with reasoning in order to handle more sophisticated learning tasks. In many cases, a reasoning task can be solved by an iterative algorithm. This algorithm is often unrolled, and used as a specialized layer in the deep architecture, which can be trained end-to-end with other neural components. Although such hybrid deep architectures have led to many empirical successes, the theoretical foundation of such architectures, especially the interplay between algorithm layers and other neural layers, remains largely unexplored. In this paper, we take an initial step towards an understanding of such hybrid deep architectures by showing that properties of the algorithm layers, such as convergence, stability, and sensitivity, are intimately related to the approximation and generalization abilities of the end-to-end model. Furthermore, our analysis matches closely our experimental observations under various conditions, suggesting that our theory can provide useful guidelines for designing deep architectures with reasoning layers.
Learning to Stop While Learning to Predict
Jun 09, 2020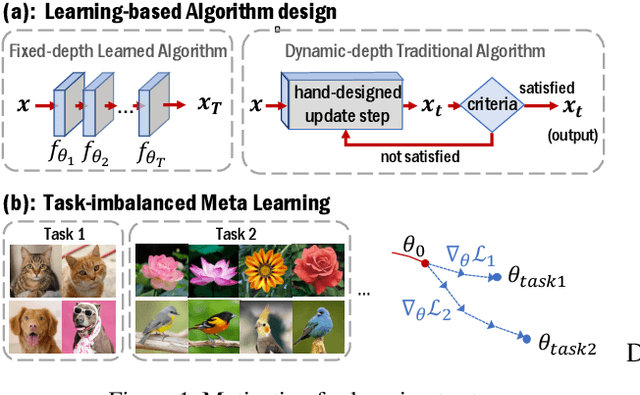

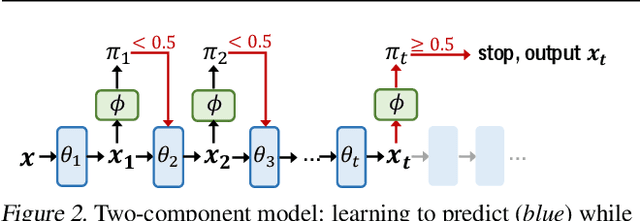
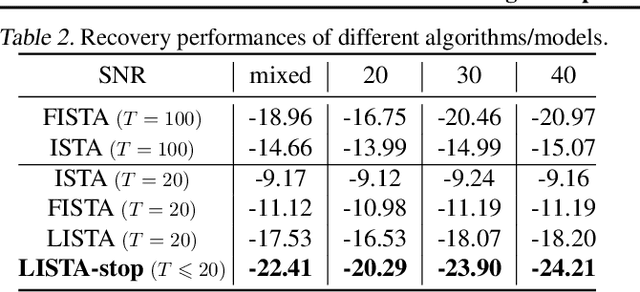
There is a recent surge of interest in designing deep architectures based on the update steps in traditional algorithms, or learning neural networks to improve and replace traditional algorithms. While traditional algorithms have certain stopping criteria for outputting results at different iterations, many algorithm-inspired deep models are restricted to a ``fixed-depth'' for all inputs. Similar to algorithms, the optimal depth of a deep architecture may be different for different input instances, either to avoid ``over-thinking'', or because we want to compute less for operations converged already. In this paper, we tackle this varying depth problem using a steerable architecture, where a feed-forward deep model and a variational stopping policy are learned together to sequentially determine the optimal number of layers for each input instance. Training such architecture is very challenging. We provide a variational Bayes perspective and design a novel and effective training procedure which decomposes the task into an oracle model learning stage and an imitation stage. Experimentally, we show that the learned deep model along with the stopping policy improves the performances on a diverse set of tasks, including learning sparse recovery, few-shot meta learning, and computer vision tasks.
RNA Secondary Structure Prediction By Learning Unrolled Algorithms
Feb 13, 2020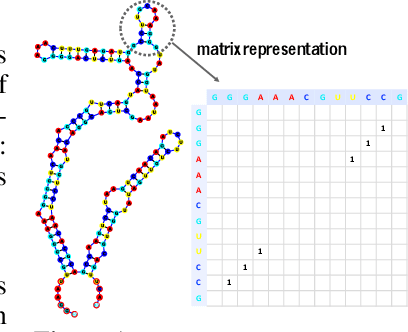
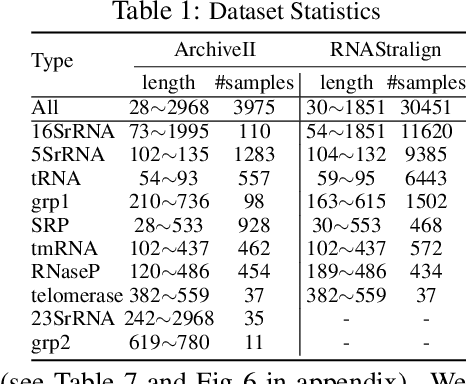
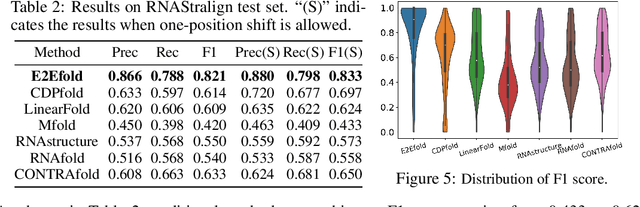
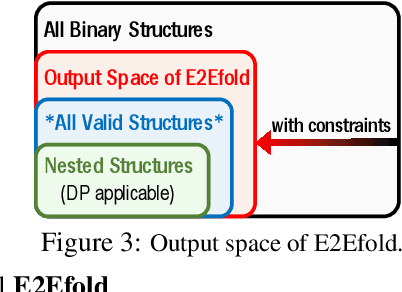
In this paper, we propose an end-to-end deep learning model, called E2Efold, for RNA secondary structure prediction which can effectively take into account the inherent constraints in the problem. The key idea of E2Efold is to directly predict the RNA base-pairing matrix, and use an unrolled algorithm for constrained programming as the template for deep architectures to enforce constraints. With comprehensive experiments on benchmark datasets, we demonstrate the superior performance of E2Efold: it predicts significantly better structures compared to previous SOTA (especially for pseudoknotted structures), while being as efficient as the fastest algorithms in terms of inference time.