 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantifying the Scanner-Induced Domain Gap in Mitosis Detection
Mar 30, 2021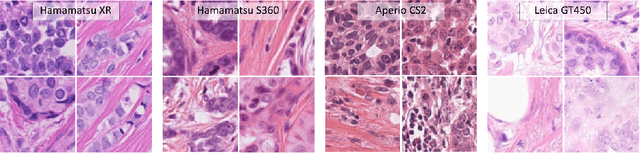

Automated detection of mitotic figures in histopathology images has seen vast improvements, thanks to modern deep learning-based pipelines. Application of these methods, however, is in practice limited by strong variability of images between labs. This results in a domain shift of the images, which causes a performance drop of the models. Hypothesizing that the scanner device plays a decisive role in this effect, we evaluated the susceptibility of a standard mitosis detection approach to the domain shift introduced by using a different whole slide scanner. Our work is based on the MICCAI-MIDOG challenge 2021 data set, which includes 200 tumor cases of human breast cancer and four scanners. Our work indicates that the domain shift induced not by biochemical variability but purely by the choice of acquisition device is underestimated so far. Models trained on images of the same scanner yielded an average F1 score of 0.683, while models trained on a single other scanner only yielded an average F1 score of 0.325. Training on another multi-domain mitosis dataset led to mean F1 scores of 0.52. We found this not to be reflected by domain-shifts measured as proxy A distance-derived metric.
Automated Scoring of Nuclear Pleomorphism Spectrum with Pathologist-level Performance in Breast Cancer
Dec 24, 2020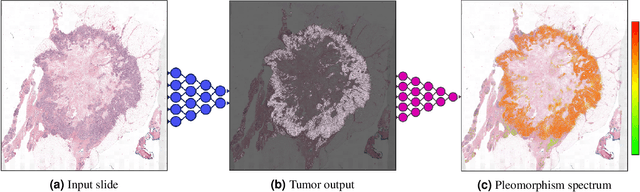
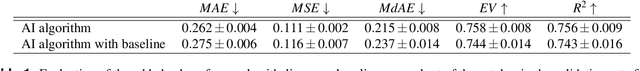
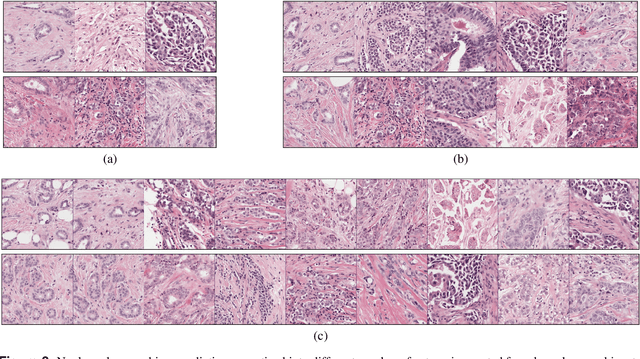
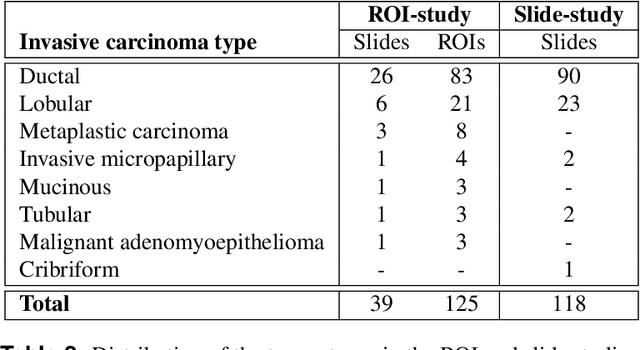
Nuclear pleomorphism, defined herein as the extent of abnormalities in the overall appearance of tumor nuclei, is one of the components of the three-tiered breast cancer grading. Given that nuclear pleomorphism reflects a continuous spectrum of variation, we trained a deep neural network on a large variety of tumor regions from the collective knowledge of several pathologists, without constraining the network to the traditional three-category classification. We also motivate an additional approach in which we discuss the additional benefit of normal epithelium as baseline, following the routine clinical practice where pathologists are trained to score nuclear pleomorphism in tumor, having the normal breast epithelium for comparison. In multiple experiments, our fully-automated approach could achieve top pathologist-level performance in select regions of interest as well as at whole slide images, compared to ten and four pathologists, respectively.
HookNet: multi-resolution convolutional neural networks for semantic segmentation in histopathology whole-slide images
Jun 22, 2020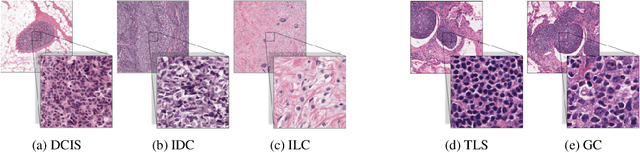
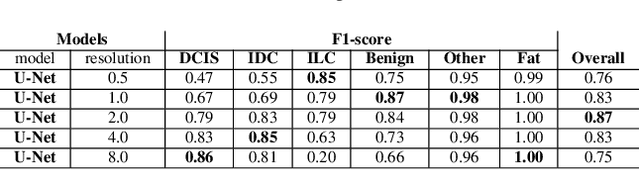
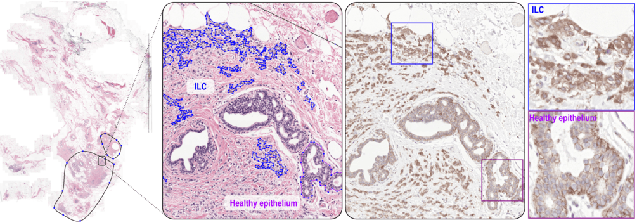
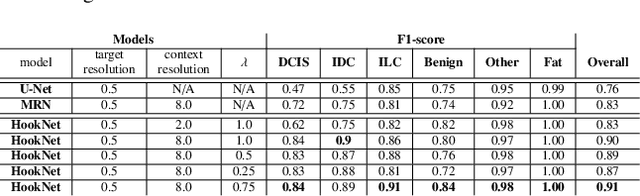
We propose HookNet, a semantic segmentation model for histopathology whole-slide images, which combines context and details via multiple branches of encoder-decoder convolutional neural networks. Concentricpatches at multiple resolutions with different fields of view are used to feed different branches of HookNet, and intermediate representations are combined via a hooking mechanism. We describe a framework to design and train HookNet for achieving high-resolution semantic segmentation and introduce constraints to guarantee pixel-wise alignment in feature maps during hooking. We show the advantages of using HookNet in two histopathology image segmentation tasks where tissue type prediction accuracy strongly depends on contextual information, namely (1) multi-class tissue segmentation in breast cancer and, (2) segmentation of tertiary lymphoid structures and germinal centers in lung cancer. Weshow the superiority of HookNet when compared with single-resolution U-Net models working at different resolutions as well as with a recently published multi-resolution model for histopathology image segmentation
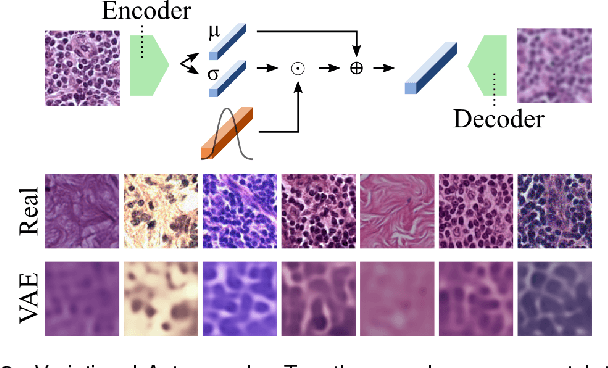
Extending Unsupervised Neural Image Compression With Supervised Multitask Learning
Apr 15, 2020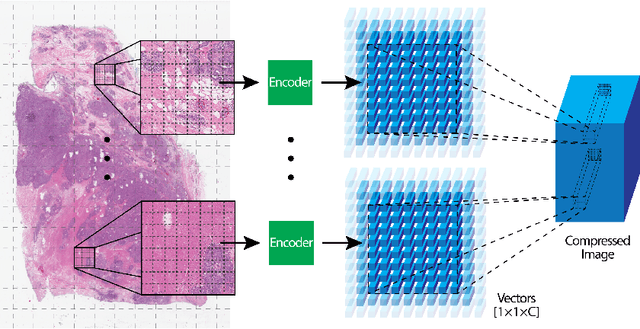
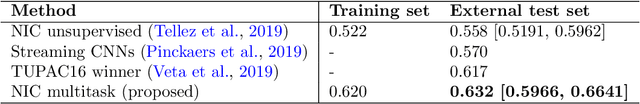
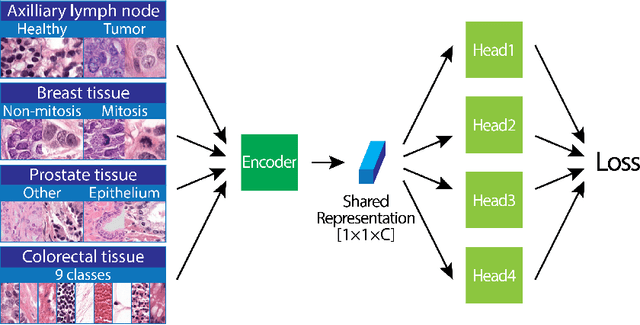
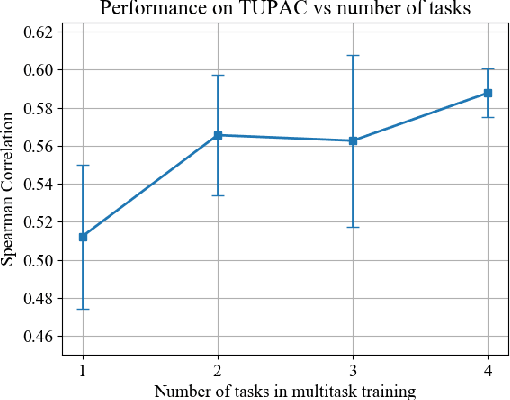
We focus on the problem of training convolutional neural networks on gigapixel histopathology images to predict image-level targets. For this purpose, we extend Neural Image Compression (NIC), an image compression framework that reduces the dimensionality of these images using an encoder network trained unsupervisedly. We propose to train this encoder using supervised multitask learning (MTL) instead. We applied the proposed MTL NIC to two histopathology datasets and three tasks. First, we obtained state-of-the-art results in the Tumor Proliferation Assessment Challenge of 2016 (TUPAC16). Second, we successfully classified histopathological growth patterns in images with colorectal liver metastasis (CLM). Third, we predicted patient risk of death by learning directly from overall survival in the same CLM data. Our experimental results suggest that the representations learned by the MTL objective are: (1) highly specific, due to the supervised training signal, and (2) transferable, since the same features perform well across different tasks. Additionally, we trained multiple encoders with different training objectives, e.g. unsupervised and variants of MTL, and observed a positive correlation between the number of tasks in MTL and the system performance on the TUPAC16 dataset.
Virtual staining for mitosis detection in Breast Histopathology
Mar 17, 2020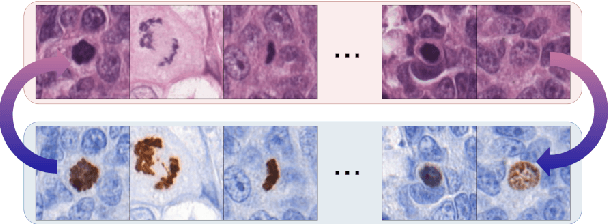
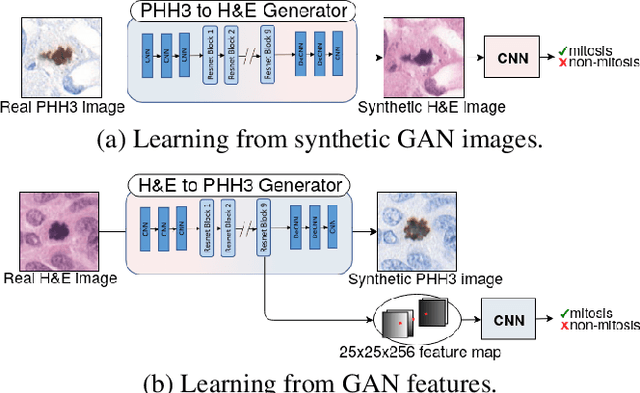
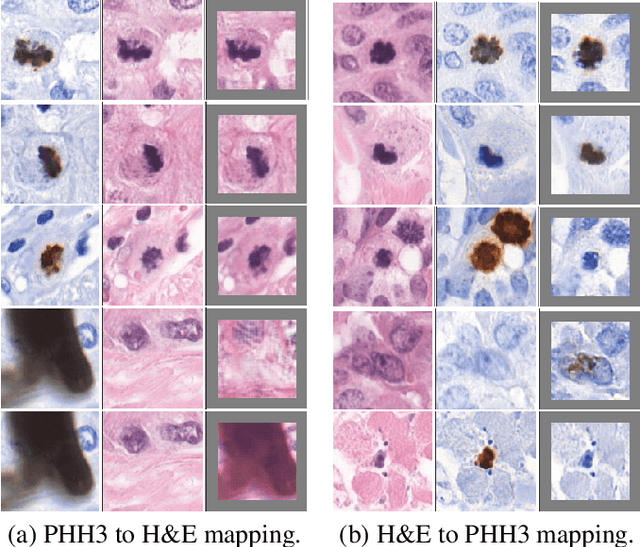
We propose a virtual staining methodology based on Generative Adversarial Networks to map histopathology images of breast cancer tissue from H&E stain to PHH3 and vice versa. We use the resulting synthetic images to build Convolutional Neural Networks (CNN) for automatic detection of mitotic figures, a strong prognostic biomarker used in routine breast cancer diagnosis and grading. We propose several scenarios, in which CNN trained with synthetically generated histopathology images perform on par with or even better than the same baseline model trained with real images. We discuss the potential of this application to scale the number of training samples without the need for manual annotations.
Needles in Haystacks: On Classifying Tiny Objects in Large Images
Aug 16, 2019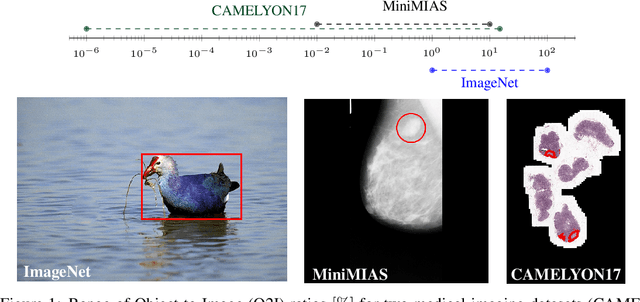
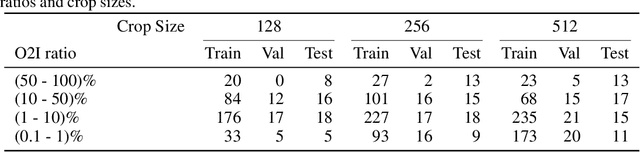
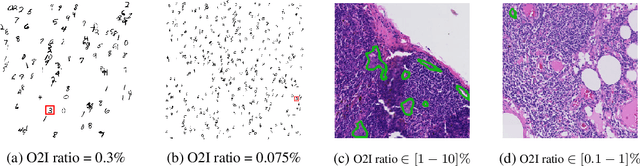
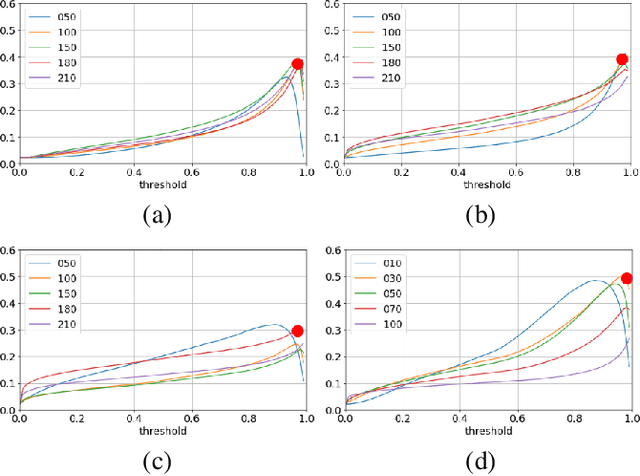
In some computer vision domains, such as medical or hyperspectral imaging, we care about the classification of tiny objects in large images. However, most Convolutional Neural Networks (CNNs) for image classification were developed and analyzed using biased datasets that contain large objects, most often, in central image positions. To assess whether classical CNN architectures work well for tiny object classification we build a comprehensive testbed containing two datasets: one derived from MNIST digits and other from histopathology images. This testbed allows us to perform controlled experiments to stress-test CNN architectures using a broad spectrum of signal-to-noise ratios. Our observations suggest that: (1) There exists a limit to signal-to-noise below which CNNs fail to generalize and that this limit is affected by dataset size - more data leading to better performances; however, the amount of training data required for the model to generalize scales rapidly with the inverse of the object-to-image ratio (2) in general, higher capacity models exhibit better generalization; (3) when knowing the approximate object sizes, adapting receptive field is beneficial; and (4) for very small signal-to-noise ratio the choice of global pooling operation affects optimization, whereas for relatively large signal-to-noise values, all tested global pooling operations exhibit similar performance.
Quantifying the effects of data augmentation and stain color normalization in convolutional neural networks for computational pathology
Feb 18, 2019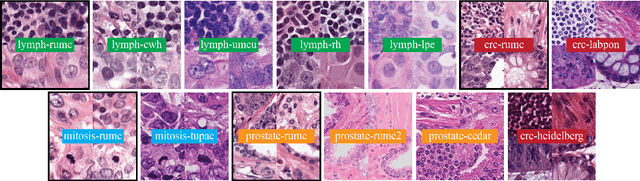
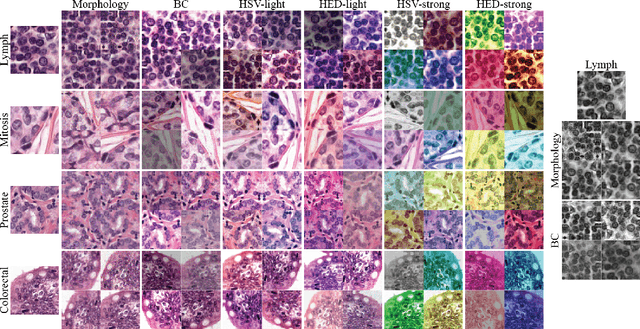
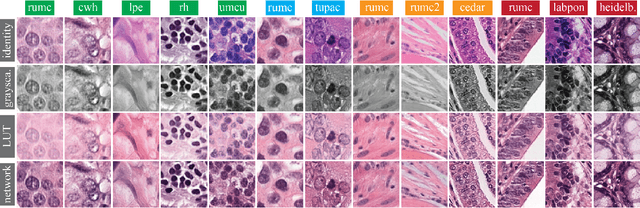
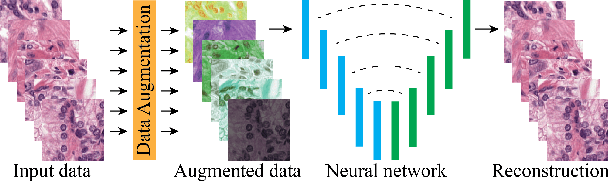
Stain variation is a phenomenon observed when distinct pathology laboratories stain tissue slides that exhibit similar but not identical color appearance. Due to this color shift between laboratories, convolutional neural networks (CNNs) trained with images from one lab often underperform on unseen images from the other lab. Several techniques have been proposed to reduce the generalization error, mainly grouped into two categories: stain color augmentation and stain color normalization. The former simulates a wide variety of realistic stain variations during training, producing stain-invariant CNNs. The latter aims to match training and test color distributions in order to reduce stain variation. For the first time, we compared some of these techniques and quantified their effect on CNN classification performance using a heterogeneous dataset of hematoxylin and eosin histopathology images from 4 organs and 9 pathology laboratories. Additionally, we propose a novel unsupervised method to perform stain color normalization using a neural network. Based on our experimental results, we provide practical guidelines on how to use stain color augmentation and stain color normalization in future computational pathology applications.
Neural Image Compression for Gigapixel Histopathology Image Analysis
Nov 07, 2018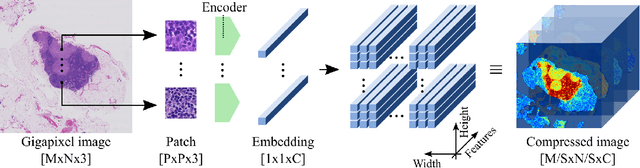
We present Neural Image Compression (NIC), a method to reduce the size of gigapixel images by mapping them to a compact latent space using neural networks. We show that this compression allows us to train convolutional neural networks on histopathology whole-slide images end-to-end using weak image-level labels.
Whole-Slide Mitosis Detection in H&E Breast Histology Using PHH3 as a Reference to Train Distilled Stain-Invariant Convolutional Networks
Aug 17, 2018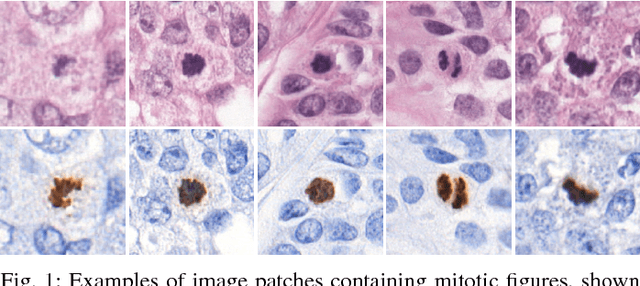
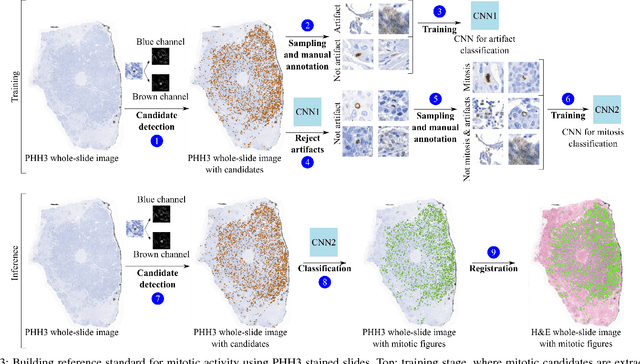
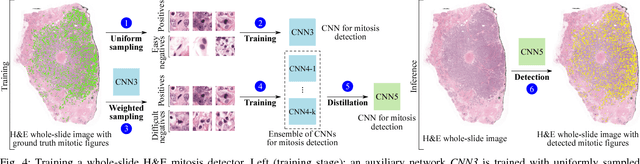
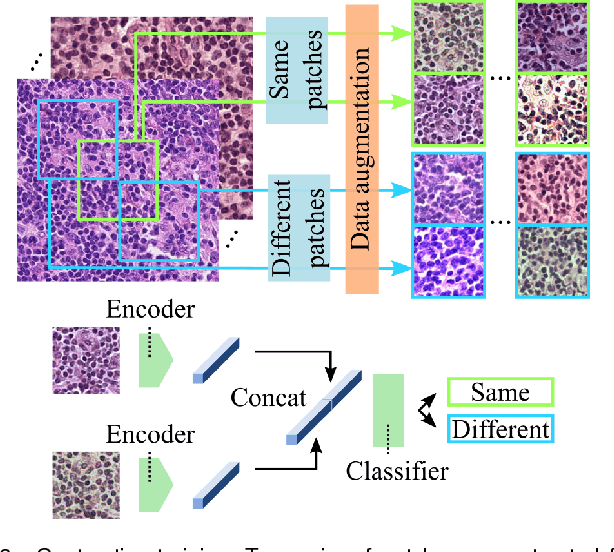
Manual counting of mitotic tumor cells in tissue sections constitutes one of the strongest prognostic markers for breast cancer. This procedure, however, is time-consuming and error-prone. We developed a method to automatically detect mitotic figures in breast cancer tissue sections based on convolutional neural networks (CNNs). Application of CNNs to hematoxylin and eosin (H&E) stained histological tissue sections is hampered by: (1) noisy and expensive reference standards established by pathologists, (2) lack of generalization due to staining variation across laboratories, and (3) high computational requirements needed to process gigapixel whole-slide images (WSIs). In this paper, we present a method to train and evaluate CNNs to specifically solve these issues in the context of mitosis detection in breast cancer WSIs. First, by combining image analysis of mitotic activity in phosphohistone-H3 (PHH3) restained slides and registration, we built a reference standard for mitosis detection in entire H&E WSIs requiring minimal manual annotation effort. Second, we designed a data augmentation strategy that creates diverse and realistic H&E stain variations by modifying the hematoxylin and eosin color channels directly. Using it during training combined with network ensembling resulted in a stain invariant mitosis detector. Third, we applied knowledge distillation to reduce the computational requirements of the mitosis detection ensemble with a negligible loss of performance. The system was trained in a single-center cohort and evaluated in an independent multicenter cohort from The Cancer Genome Atlas on the three tasks of the Tumor Proliferation Assessment Challenge (TUPAC). We obtained a performance within the top-3 best methods for most of the tasks of the challenge.
Predicting breast tumor proliferation from whole-slide images: the TUPAC16 challenge
Jul 22, 2018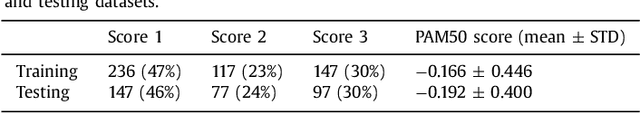
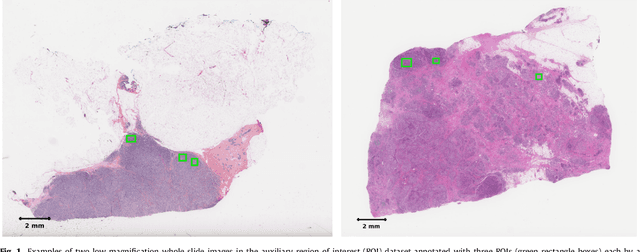
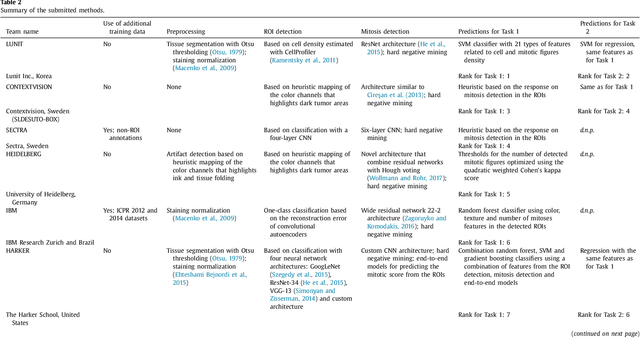
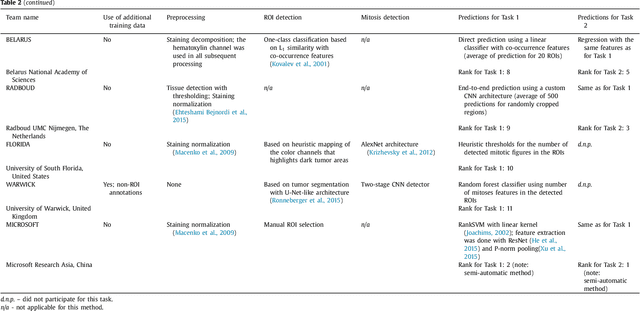
Tumor proliferation is an important biomarker indicative of the prognosis of breast cancer patients. Assessment of tumor proliferation in a clinical setting is highly subjective and labor-intensive task. Previous efforts to automate tumor proliferation assessment by image analysis only focused on mitosis detection in predefined tumor regions. However, in a real-world scenario, automatic mitosis detection should be performed in whole-slide images (WSIs) and an automatic method should be able to produce a tumor proliferation score given a WSI as input. To address this, we organized the TUmor Proliferation Assessment Challenge 2016 (TUPAC16) on prediction of tumor proliferation scores from WSIs. The challenge dataset consisted of 500 training and 321 testing breast cancer histopathology WSIs. In order to ensure fair and independent evaluation, only the ground truth for the training dataset was provided to the challenge participants. The first task of the challenge was to predict mitotic scores, i.e., to reproduce the manual method of assessing tumor proliferation by a pathologist. The second task was to predict the gene expression based PAM50 proliferation scores from the WSI. The best performing automatic method for the first task achieved a quadratic-weighted Cohen's kappa score of $\kappa$ = 0.567, 95% CI [0.464, 0.671] between the predicted scores and the ground truth. For the second task, the predictions of the top method had a Spearman's correlation coefficient of r = 0.617, 95% CI [0.581 0.651] with the ground truth. This was the first study that investigated tumor proliferation assessment from WSIs. The achieved results are promising given the difficulty of the tasks and weakly-labelled nature of the ground truth. However, further research is needed to improve the practical utility of image analysis methods for this task.