 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWhole-Slide Mitosis Detection in H&E Breast Histology Using PHH3 as a Reference to Train Distilled Stain-Invariant Convolutional Networks
Aug 17, 2018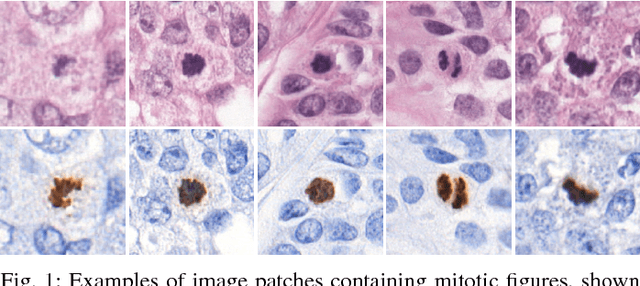
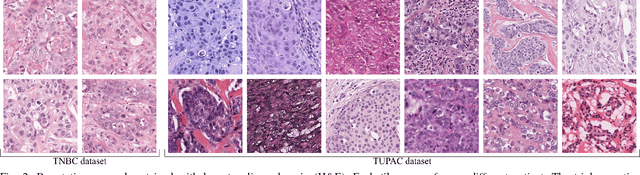
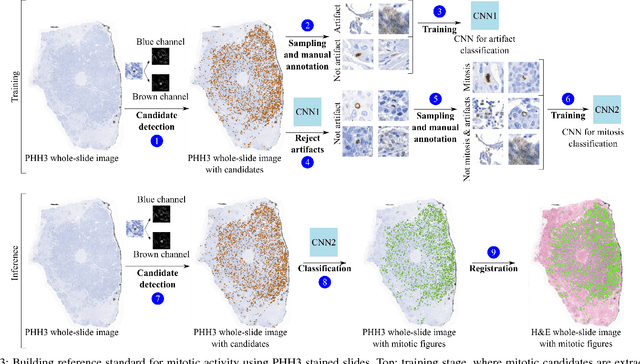
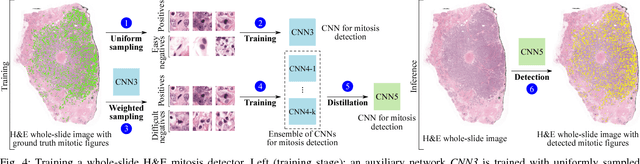
Manual counting of mitotic tumor cells in tissue sections constitutes one of the strongest prognostic markers for breast cancer. This procedure, however, is time-consuming and error-prone. We developed a method to automatically detect mitotic figures in breast cancer tissue sections based on convolutional neural networks (CNNs). Application of CNNs to hematoxylin and eosin (H&E) stained histological tissue sections is hampered by: (1) noisy and expensive reference standards established by pathologists, (2) lack of generalization due to staining variation across laboratories, and (3) high computational requirements needed to process gigapixel whole-slide images (WSIs). In this paper, we present a method to train and evaluate CNNs to specifically solve these issues in the context of mitosis detection in breast cancer WSIs. First, by combining image analysis of mitotic activity in phosphohistone-H3 (PHH3) restained slides and registration, we built a reference standard for mitosis detection in entire H&E WSIs requiring minimal manual annotation effort. Second, we designed a data augmentation strategy that creates diverse and realistic H&E stain variations by modifying the hematoxylin and eosin color channels directly. Using it during training combined with network ensembling resulted in a stain invariant mitosis detector. Third, we applied knowledge distillation to reduce the computational requirements of the mitosis detection ensemble with a negligible loss of performance. The system was trained in a single-center cohort and evaluated in an independent multicenter cohort from The Cancer Genome Atlas on the three tasks of the Tumor Proliferation Assessment Challenge (TUPAC). We obtained a performance within the top-3 best methods for most of the tasks of the challenge.
Epithelium segmentation using deep learning in H&E-stained prostate specimens with immunohistochemistry as reference standard
Aug 17, 2018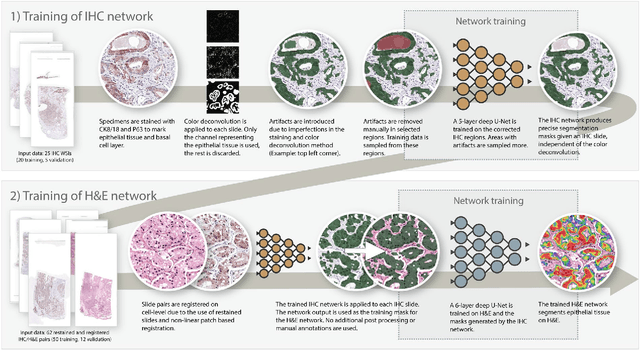
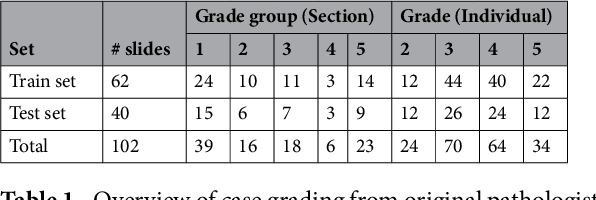
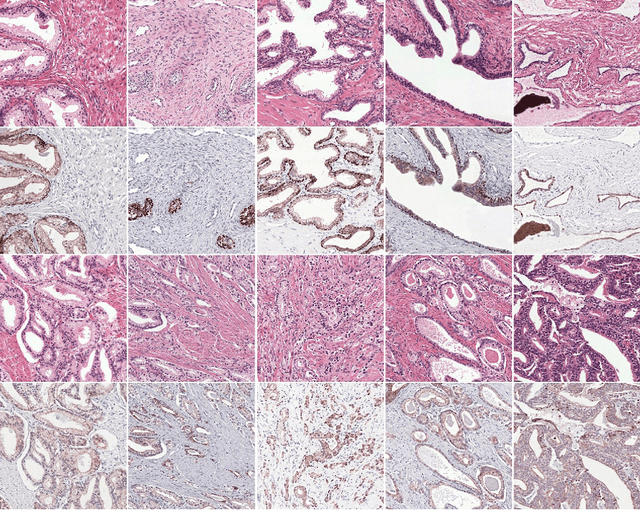
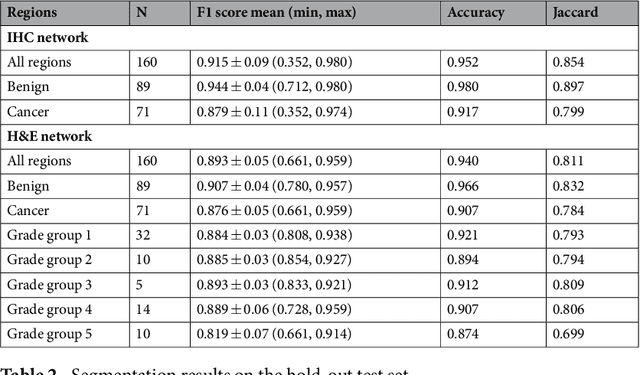
Prostate cancer (PCa) is graded by pathologists by examining the architectural pattern of cancerous epithelial tissue on hematoxylin and eosin (H&E) stained slides. Given the importance of gland morphology, automatically differentiating between glandular epithelial tissue and other tissues is an important prerequisite for the development of automated methods for detecting PCa. We propose a new method, using deep learning, for automatically segmenting epithelial tissue in digitized prostatectomy slides. We employed immunohistochemistry (IHC) to render the ground truth less subjective and more precise compared to manual outlining on H&E slides, especially in areas with high-grade and poorly differentiated PCa. Our dataset consisted of 102 tissue blocks, including both low and high grade PCa. From each block a single new section was cut, stained with H&E, scanned, restained using P63 and CK8/18 to highlight the epithelial structure, and scanned again. The H&E slides were co-registered to the IHC slides. On a subset of the IHC slides we applied color deconvolution, corrected stain errors manually, and trained a U-Net to perform segmentation of epithelial structures. Whole-slide segmentation masks generated by the IHC U-Net were used to train a second U-Net on H&E. Our system makes precise cell-level segmentations and segments both intact glands as well as individual (tumor) epithelial cells. We achieved an F1-score of 0.895 on a hold-out test set and 0.827 on an external reference set from a different center. We envision this segmentation as being the first part of a fully automated prostate cancer detection and grading pipeline.
Comparison of Different Methods for Tissue Segmentation in Histopathological Whole-Slide Images
Apr 03, 2017
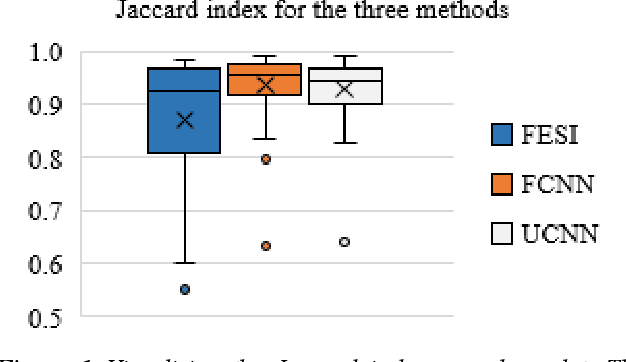


Tissue segmentation is an important pre-requisite for efficient and accurate diagnostics in digital pathology. However, it is well known that whole-slide scanners can fail in detecting all tissue regions, for example due to the tissue type, or due to weak staining because their tissue detection algorithms are not robust enough. In this paper, we introduce two different convolutional neural network architectures for whole slide image segmentation to accurately identify the tissue sections. We also compare the algorithms to a published traditional method. We collected 54 whole slide images with differing stains and tissue types from three laboratories to validate our algorithms. We show that while the two methods do not differ significantly they outperform their traditional counterpart (Jaccard index of 0.937 and 0.929 vs. 0.870, p < 0.01).