 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCORE - A Cell-Level Coarse-to-Fine Image Registration Engine for Multi-stain Image Alignment
Nov 12, 2025Accurate and efficient registration of whole slide images (WSIs) is essential for high-resolution, nuclei-level analysis in multi-stained tissue slides. We propose a novel coarse-to-fine framework CORE for accurate nuclei-level registration across diverse multimodal whole-slide image (WSI) datasets. The coarse registration stage leverages prompt-based tissue mask extraction to effectively filter out artefacts and non-tissue regions, followed by global alignment using tissue morphology and ac- celerated dense feature matching with a pre-trained feature extractor. From the coarsely aligned slides, nuclei centroids are detected and subjected to fine-grained rigid registration using a custom, shape-aware point-set registration model. Finally, non-rigid alignment at the cellular level is achieved by estimating a non-linear dis- placement field using Coherent Point Drift (CPD). Our approach benefits from automatically generated nuclei that enhance the accuracy of deformable registra- tion and ensure precise nuclei-level correspondence across modalities. The pro- posed model is evaluated on three publicly available WSI registration datasets, and two private datasets. We show that CORE outperforms current state-of-the-art methods in terms of generalisability, precision, and robustness in bright-field and immunofluorescence microscopy WSIs
The case for delegated AI autonomy for Human AI teaming in healthcare
Mar 24, 2025In this paper we propose an advanced approach to integrating artificial intelligence (AI) into healthcare: autonomous decision support. This approach allows the AI algorithm to act autonomously for a subset of patient cases whilst serving a supportive role in other subsets of patient cases based on defined delegation criteria. By leveraging the complementary strengths of both humans and AI, it aims to deliver greater overall performance than existing human-AI teaming models. It ensures safe handling of patient cases and potentially reduces clinician review time, whilst being mindful of AI tool limitations. After setting the approach within the context of current human-AI teaming models, we outline the delegation criteria and apply them to a specific AI-based tool used in histopathology. The potential impact of the approach and the regulatory requirements for its successful implementation are then discussed.
CellOMaps: A Compact Representation for Robust Classification of Lung Adenocarcinoma Growth Patterns
Jan 14, 2025



Lung adenocarcinoma (LUAD) is a morphologically heterogeneous disease, characterized by five primary histological growth patterns. The classification of such patterns is crucial due to their direct relation to prognosis but the high subjectivity and observer variability pose a major challenge. Although several studies have developed machine learning methods for growth pattern classification, they either only report the predominant pattern per slide or lack proper evaluation. We propose a generalizable machine learning pipeline capable of classifying lung tissue into one of the five patterns or as non-tumor. The proposed pipeline's strength lies in a novel compact Cell Organization Maps (cellOMaps) representation that captures the cellular spatial patterns from Hematoxylin and Eosin whole slide images (WSIs). The proposed pipeline provides state-of-the-art performance on LUAD growth pattern classification when evaluated on both internal unseen slides and external datasets, significantly outperforming the current approaches. In addition, our preliminary results show that the model's outputs can be used to predict patients Tumor Mutational Burden (TMB) levels.
Large Multimodal Model based Standardisation of Pathology Reports with Confidence and their Prognostic Significance
May 03, 2024



Pathology reports are rich in clinical and pathological details but are often presented in free-text format. The unstructured nature of these reports presents a significant challenge limiting the accessibility of their content. In this work, we present a practical approach based on the use of large multimodal models (LMMs) for automatically extracting information from scanned images of pathology reports with the goal of generating a standardised report specifying the value of different fields along with estimated confidence about the accuracy of the extracted fields. The proposed approach overcomes limitations of existing methods which do not assign confidence scores to extracted fields limiting their practical use. The proposed framework uses two stages of prompting a Large Multimodal Model (LMM) for information extraction and validation. The framework generalises to textual reports from multiple medical centres as well as scanned images of legacy pathology reports. We show that the estimated confidence is an effective indicator of the accuracy of the extracted information that can be used to select only accurately extracted fields. We also show the prognostic significance of structured and unstructured data from pathology reports and show that the automatically extracted field values significant prognostic value for patient stratification. The framework is available for evaluation via the URL: https://labieb.dcs.warwick.ac.uk/.
CoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.
One Model is All You Need: Multi-Task Learning Enables Simultaneous Histology Image Segmentation and Classification
Feb 28, 2022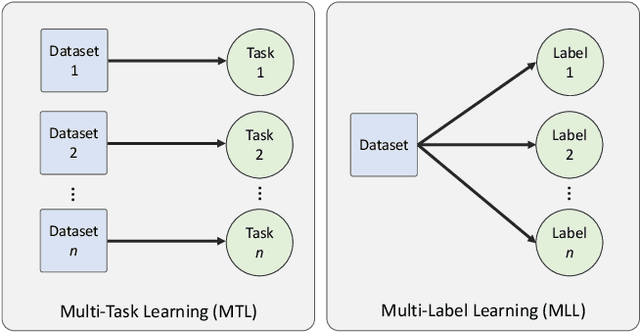
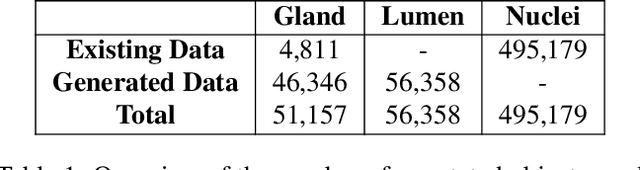
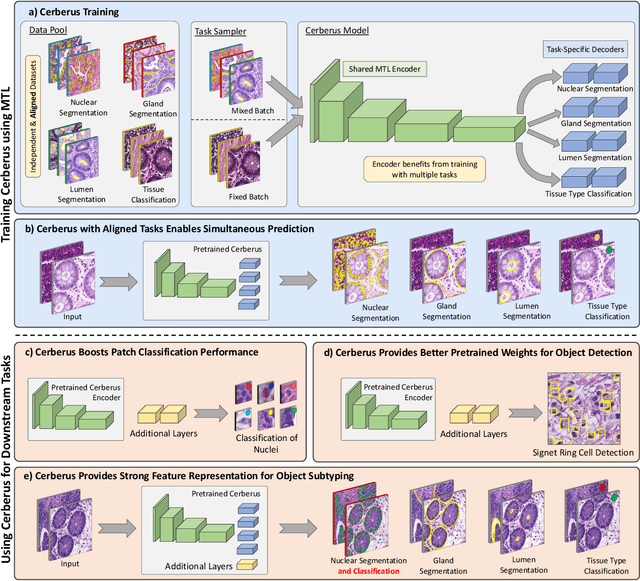
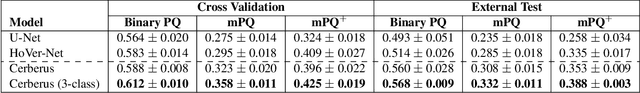
The recent surge in performance for image analysis of digitised pathology slides can largely be attributed to the advance of deep learning. Deep models can be used to initially localise various structures in the tissue and hence facilitate the extraction of interpretable features for biomarker discovery. However, these models are typically trained for a single task and therefore scale poorly as we wish to adapt the model for an increasing number of different tasks. Also, supervised deep learning models are very data hungry and therefore rely on large amounts of training data to perform well. In this paper we present a multi-task learning approach for segmentation and classification of nuclei, glands, lumen and different tissue regions that leverages data from multiple independent data sources. While ensuring that our tasks are aligned by the same tissue type and resolution, we enable simultaneous prediction with a single network. As a result of feature sharing, we also show that the learned representation can be used to improve downstream tasks, including nuclear classification and signet ring cell detection. As part of this work, we use a large dataset consisting of over 600K objects for segmentation and 440K patches for classification and make the data publicly available. We use our approach to process the colorectal subset of TCGA, consisting of 599 whole-slide images, to localise 377 million, 900K and 2.1 million nuclei, glands and lumen respectively. We make this resource available to remove a major barrier in the development of explainable models for computational pathology.
Deep Learning based Prediction of MSI in Colorectal Cancer via Prediction of the Status of MMR Markers
Feb 24, 2022

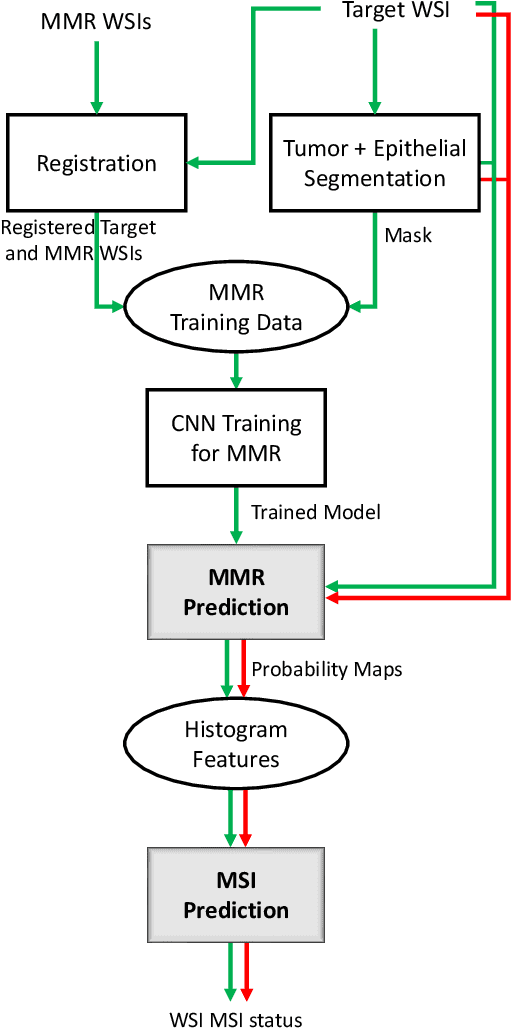
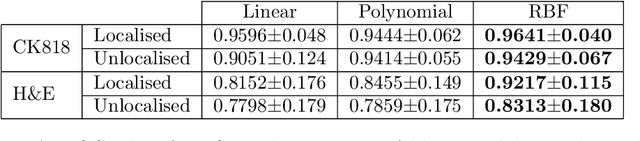
An accurate diagnosis and profiling of tumour are critical to the best treatment choices for cancer patients. In addition to the cancer type and its aggressiveness, molecular heterogeneity also plays a vital role in treatment selection. MSI or MMR deficiency is one of the well-studied aberrations in terms of molecular changes. Colorectal cancer patients with MMR deficiency respond well to immunotherapy, hence assessment of the relevant molecular markers can assist clinicians in making optimal treatment selections for patients. Immunohistochemistry is one of the ways for identifying these molecular changes which requires additional sections of tumour tissue. Introduction of automated methods that can predict MSI or MMR status from a target image without the need for additional sections can substantially reduce the cost associated with it. In this work, we present our work on predicting MSI status in a two-stage process using a single target slide either stained with CK818 or H\&E. First, we train a multi-headed convolutional neural network model where each head is responsible for predicting one of the MMR protein expressions. To this end, we perform registration of MMR slides to the target slide as a pre-processing step. In the second stage, statistical features computed from the MMR prediction maps are used for the final MSI prediction. Our results demonstrate that MSI classification can be improved on incorporating fine-grained MMR labels in comparison to the previous approaches in which coarse labels (MSI/MSS) are utilised.
Towards Launching AI Algorithms for Cellular Pathology into Clinical & Pharmaceutical Orbits
Dec 17, 2021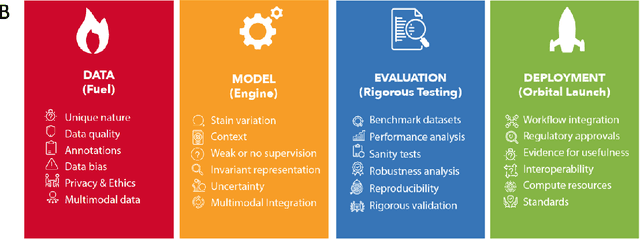

Computational Pathology (CPath) is an emerging field concerned with the study of tissue pathology via computational algorithms for the processing and analysis of digitized high-resolution images of tissue slides. Recent deep learning based developments in CPath have successfully leveraged sheer volume of raw pixel data in histology images for predicting target parameters in the domains of diagnostics, prognostics, treatment sensitivity and patient stratification -- heralding the promise of a new data-driven AI era for both histopathology and oncology. With data serving as the fuel and AI as the engine, CPath algorithms are poised to be ready for takeoff and eventual launch into clinical and pharmaceutical orbits. In this paper, we discuss CPath limitations and associated challenges to enable the readers distinguish hope from hype and provide directions for future research to overcome some of the major challenges faced by this budding field to enable its launch into the two orbits.
CoNIC: Colon Nuclei Identification and Counting Challenge 2022
Nov 29, 2021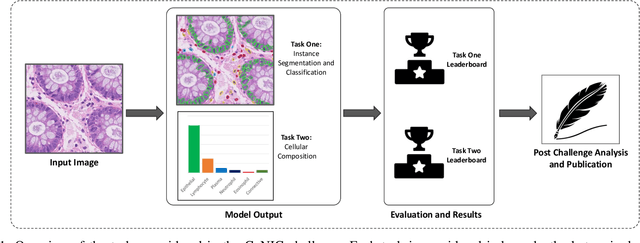
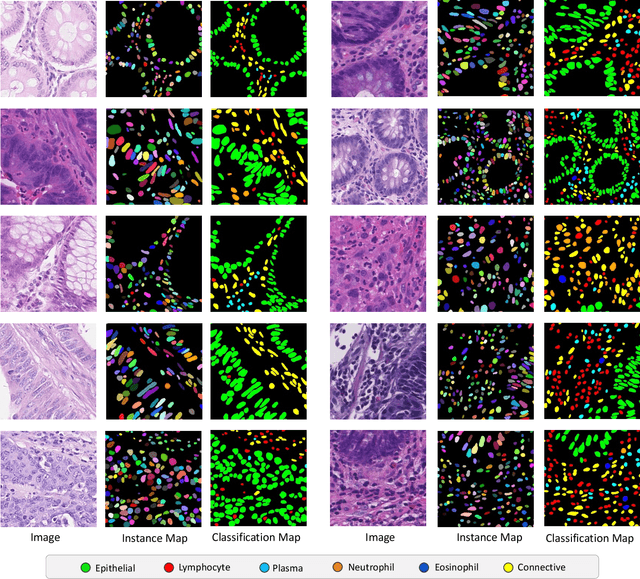
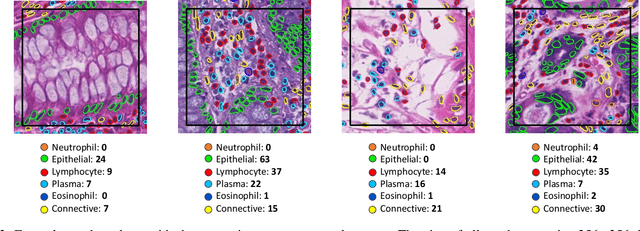
Nuclear segmentation, classification and quantification within Haematoxylin & Eosin stained histology images enables the extraction of interpretable cell-based features that can be used in downstream explainable models in computational pathology (CPath). However, automatic recognition of different nuclei is faced with a major challenge in that there are several different types of nuclei, some of them exhibiting large intra-class variability. To help drive forward research and innovation for automatic nuclei recognition in CPath, we organise the Colon Nuclei Identification and Counting (CoNIC) Challenge. The challenge encourages researchers to develop algorithms that perform segmentation, classification and counting of nuclei within the current largest known publicly available nuclei-level dataset in CPath, containing around half a million labelled nuclei. Therefore, the CoNIC challenge utilises over 10 times the number of nuclei as the previous largest challenge dataset for nuclei recognition. It is important for algorithms to be robust to input variation if we wish to deploy them in a clinical setting. Therefore, as part of this challenge we will also test the sensitivity of each submitted algorithm to certain input variations.
Lizard: A Large-Scale Dataset for Colonic Nuclear Instance Segmentation and Classification
Aug 25, 2021
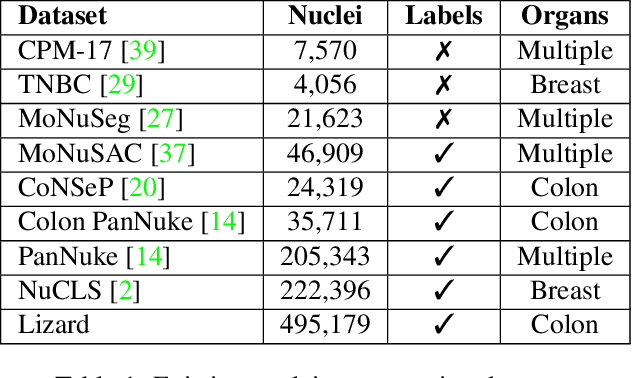
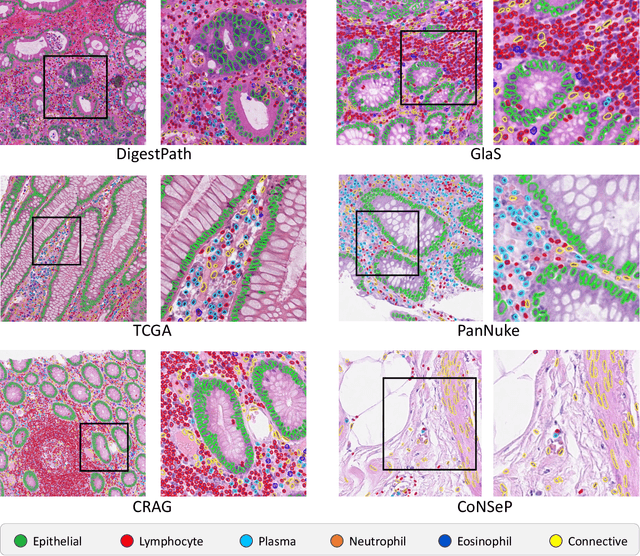
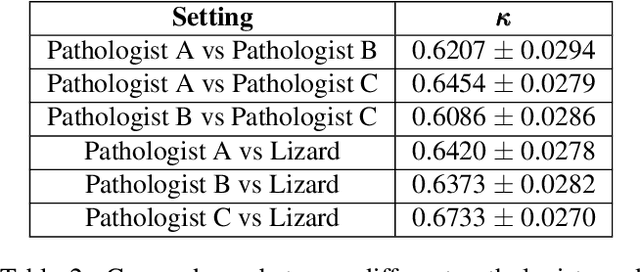
The development of deep segmentation models for computational pathology (CPath) can help foster the investigation of interpretable morphological biomarkers. Yet, there is a major bottleneck in the success of such approaches because supervised deep learning models require an abundance of accurately labelled data. This issue is exacerbated in the field of CPath because the generation of detailed annotations usually demands the input of a pathologist to be able to distinguish between different tissue constructs and nuclei. Manually labelling nuclei may not be a feasible approach for collecting large-scale annotated datasets, especially when a single image region can contain thousands of different cells. However, solely relying on automatic generation of annotations will limit the accuracy and reliability of ground truth. Therefore, to help overcome the above challenges, we propose a multi-stage annotation pipeline to enable the collection of large-scale datasets for histology image analysis, with pathologist-in-the-loop refinement steps. Using this pipeline, we generate the largest known nuclear instance segmentation and classification dataset, containing nearly half a million labelled nuclei in H&E stained colon tissue. We have released the dataset and encourage the research community to utilise it to drive forward the development of downstream cell-based models in CPath.