 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCellOMaps: A Compact Representation for Robust Classification of Lung Adenocarcinoma Growth Patterns
Jan 14, 2025



Lung adenocarcinoma (LUAD) is a morphologically heterogeneous disease, characterized by five primary histological growth patterns. The classification of such patterns is crucial due to their direct relation to prognosis but the high subjectivity and observer variability pose a major challenge. Although several studies have developed machine learning methods for growth pattern classification, they either only report the predominant pattern per slide or lack proper evaluation. We propose a generalizable machine learning pipeline capable of classifying lung tissue into one of the five patterns or as non-tumor. The proposed pipeline's strength lies in a novel compact Cell Organization Maps (cellOMaps) representation that captures the cellular spatial patterns from Hematoxylin and Eosin whole slide images (WSIs). The proposed pipeline provides state-of-the-art performance on LUAD growth pattern classification when evaluated on both internal unseen slides and external datasets, significantly outperforming the current approaches. In addition, our preliminary results show that the model's outputs can be used to predict patients Tumor Mutational Burden (TMB) levels.
Cell Maps Representation For Lung Adenocarcinoma Growth Patterns Classification In Whole Slide Images
Nov 27, 2023


Lung adenocarcinoma is a morphologically heterogeneous disease, characterized by five primary histologic growth patterns. The quantity of these patterns can be related to tumor behavior and has a significant impact on patient prognosis. In this work, we propose a novel machine learning pipeline capable of classifying tissue tiles into one of the five patterns or as non-tumor, with an Area Under the Receiver Operating Characteristic Curve (AUCROC) score of 0.97. Our model's strength lies in its comprehensive consideration of cellular spatial patterns, where it first generates cell maps from Hematoxylin and Eosin (H&E) whole slide images (WSIs), which are then fed into a convolutional neural network classification model. Exploiting these cell maps provides the model with robust generalizability to new data, achieving approximately 30% higher accuracy on unseen test-sets compared to current state of the art approaches. The insights derived from our model can be used to predict prognosis, enhancing patient outcomes.
Lizard: A Large-Scale Dataset for Colonic Nuclear Instance Segmentation and Classification
Aug 25, 2021
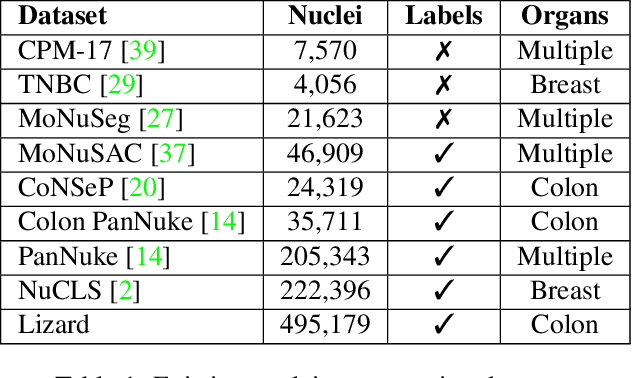
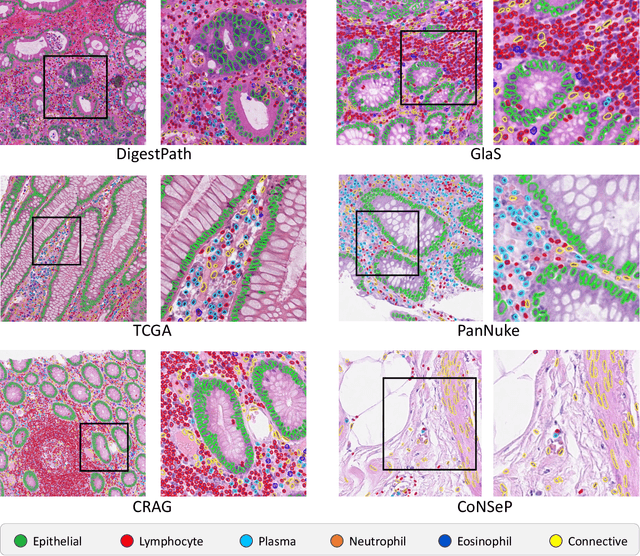
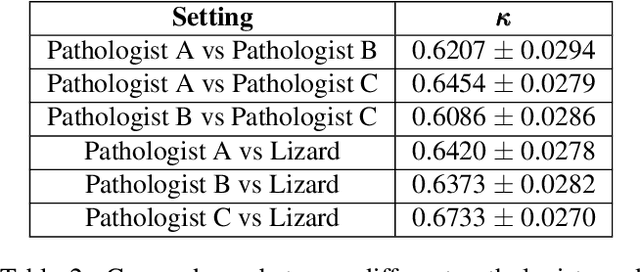
The development of deep segmentation models for computational pathology (CPath) can help foster the investigation of interpretable morphological biomarkers. Yet, there is a major bottleneck in the success of such approaches because supervised deep learning models require an abundance of accurately labelled data. This issue is exacerbated in the field of CPath because the generation of detailed annotations usually demands the input of a pathologist to be able to distinguish between different tissue constructs and nuclei. Manually labelling nuclei may not be a feasible approach for collecting large-scale annotated datasets, especially when a single image region can contain thousands of different cells. However, solely relying on automatic generation of annotations will limit the accuracy and reliability of ground truth. Therefore, to help overcome the above challenges, we propose a multi-stage annotation pipeline to enable the collection of large-scale datasets for histology image analysis, with pathologist-in-the-loop refinement steps. Using this pipeline, we generate the largest known nuclear instance segmentation and classification dataset, containing nearly half a million labelled nuclei in H&E stained colon tissue. We have released the dataset and encourage the research community to utilise it to drive forward the development of downstream cell-based models in CPath.
Now You See It, Now You Dont: Adversarial Vulnerabilities in Computational Pathology
Jun 16, 2021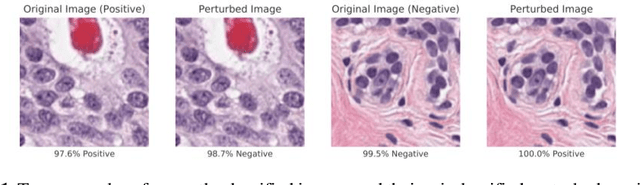

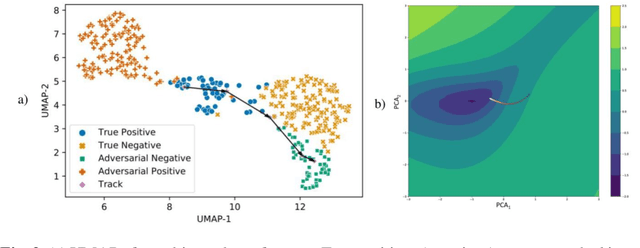
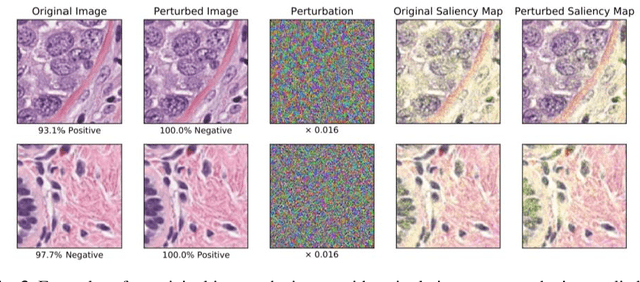
Deep learning models are routinely employed in computational pathology (CPath) for solving problems of diagnostic and prognostic significance. Typically, the generalization performance of CPath models is analyzed using evaluation protocols such as cross-validation and testing on multi-centric cohorts. However, to ensure that such CPath solutions are robust and safe for use in a clinical setting, a critical analysis of their predictive performance and vulnerability to adversarial attacks is required, which is the focus of this paper. Specifically, we show that a highly accurate model for classification of tumour patches in pathology images (AUC > 0.95) can easily be attacked with minimal perturbations which are imperceptible to lay humans and trained pathologists alike. Our analytical results show that it is possible to generate single-instance white-box attacks on specific input images with high success rate and low perturbation energy. Furthermore, we have also generated a single universal perturbation matrix using the training dataset only which, when added to unseen test images, results in forcing the trained neural network to flip its prediction labels with high confidence at a success rate of > 84%. We systematically analyze the relationship between perturbation energy of an adversarial attack, its impact on morphological constructs of clinical significance, their perceptibility by a trained pathologist and saliency maps obtained using deep learning models. Based on our analysis, we strongly recommend that computational pathology models be critically analyzed using the proposed adversarial validation strategy prior to clinical adoption.
PanNuke Dataset Extension, Insights and Baselines
Apr 22, 2020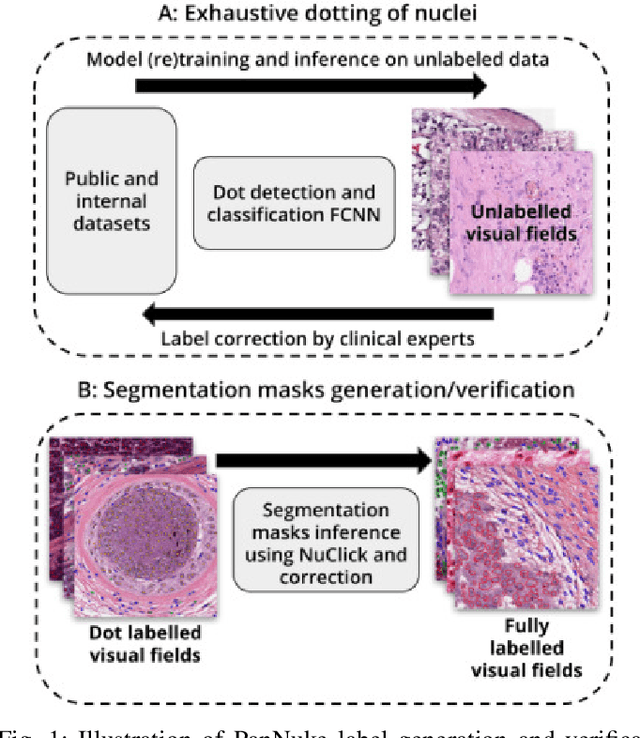
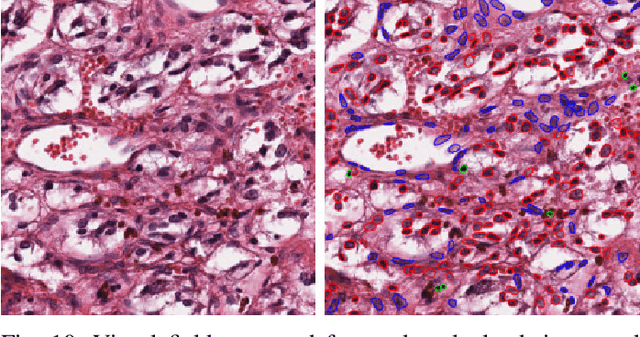
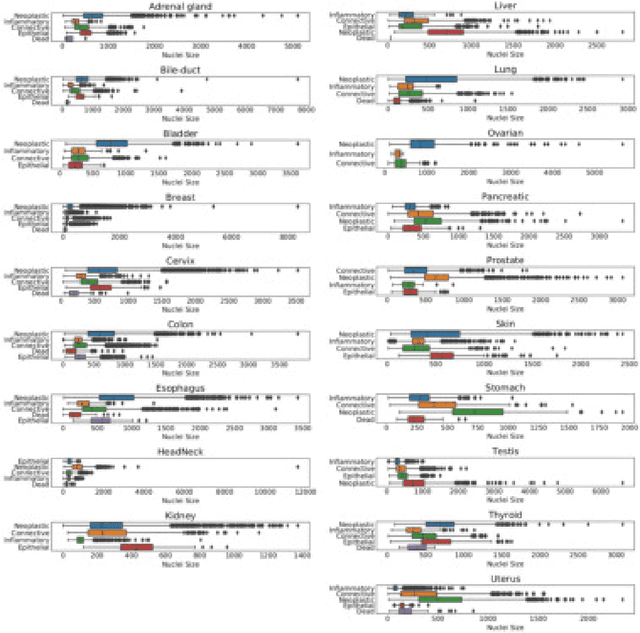
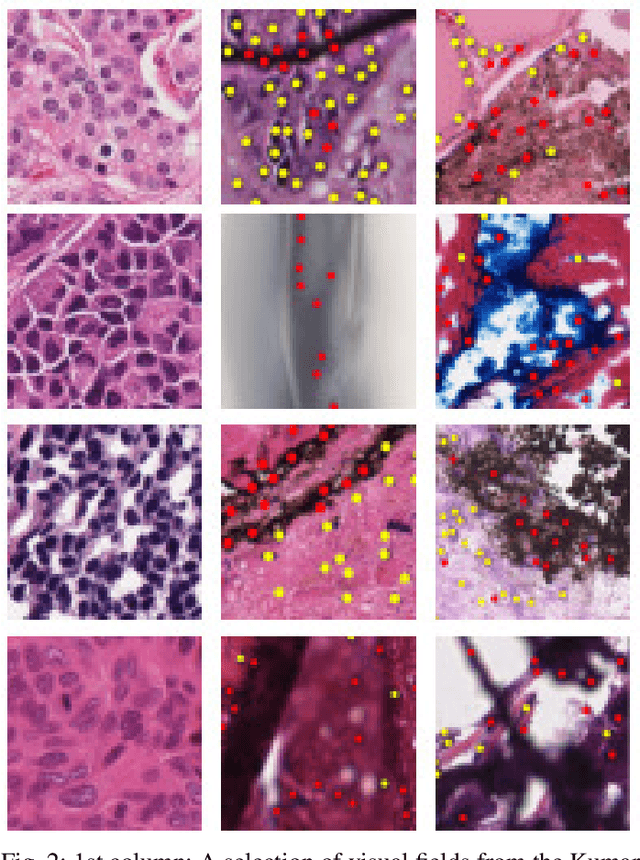
The emerging area of computational pathology (CPath) is ripe ground for the application of deep learning (DL) methods to healthcare due to the sheer volume of raw pixel data in whole-slide images (WSIs) of cancerous tissue slides. However, it is imperative for the DL algorithms relying on nuclei-level details to be able to cope with data from `the clinical wild', which tends to be quite challenging. We study, and extend recently released PanNuke dataset consisting of ~200,000 nuclei categorized into 5 clinically important classes for the challenging tasks of segmenting and classifying nuclei in WSIs. Previous pan-cancer datasets consisted of only up to 9 different tissues and up to 21,000 unlabeled nuclei and just over 24,000 labeled nuclei with segmentation masks. PanNuke consists of 19 different tissue types that have been semi-automatically annotated and quality controlled by clinical pathologists, leading to a dataset with statistics similar to the clinical wild and with minimal selection bias. We study the performance of segmentation and classification models when applied to the proposed dataset and demonstrate the application of models trained on PanNuke to whole-slide images. We provide comprehensive statistics about the dataset and outline recommendations and research directions to address the limitations of existing DL tools when applied to real-world CPath applications.
Context-Aware Convolutional Neural Network for Grading of Colorectal Cancer Histology Images
Jul 22, 2019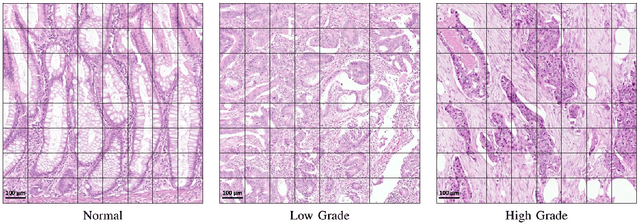
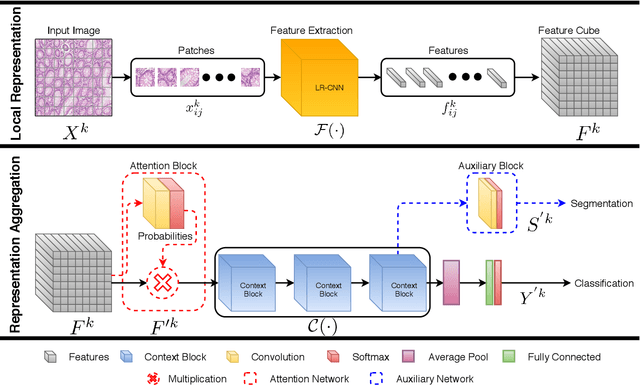
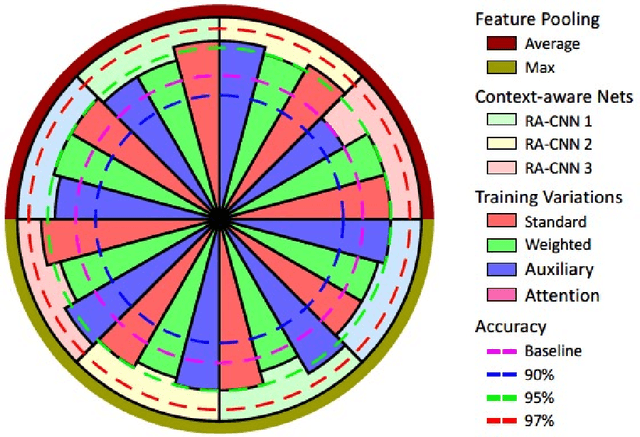

Digital histology images are amenable to the application of convolutional neural network (CNN) for analysis due to the sheer size of pixel data present in them. CNNs are generally used for representation learning from small image patches (e.g. 224x224) extracted from digital histology images due to computational and memory constraints. However, this approach does not incorporate high-resolution contextual information in histology images. We propose a novel way to incorporate larger context by a context-aware neural network based on images with a dimension of 1,792x1,792 pixels. The proposed framework first encodes the local representation of a histology image into high dimensional features then aggregates the features by considering their spatial organization to make a final prediction. The proposed method is evaluated for colorectal cancer grading and breast cancer classification. A comprehensive analysis of some variants of the proposed method is presented. Our method outperformed the traditional patch-based approaches, problem-specific methods, and existing context-based methods quantitatively by a margin of 3.61%. Code and dataset related information is available at this link: https://tia-lab.github.io/Context-Aware-CNN