 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeX Ray Diffraction
Papers and Code
Machine Learning-Driven Crystal System Prediction for Perovskites Using Augmented X-ray Diffraction Data
Feb 04, 2026Prediction of crystal system from X-ray diffraction (XRD) spectra is a critical task in materials science, particularly for perovskite materials which are known for their diverse applications in photovoltaics, optoelectronics, and catalysis. In this study, we present a machine learning (ML)-driven framework that leverages advanced models, including Time Series Forest (TSF), Random Forest (RF), Extreme Gradient Boosting (XGBoost), Recurrent Neural Network (RNN), Long Short-Term Memory (LSTM), Gated Recurrent Unit (GRU), and a simple feedforward neural network (NN), to classify crystal systems, point groups, and space groups from XRD data of perovskite materials. To address class imbalance and enhance model robustness, we integrated feature augmentation strategies such as Synthetic Minority Over-sampling Technique (SMOTE), class weighting, jittering, and spectrum shifting, along with efficient data preprocessing pipelines. The TSF model with SMOTE augmentation achieved strong performance for crystal system prediction, with a Matthews correlation coefficient (MCC) of 0.9, an F1 score of 0.92, and an accuracy of 97.76%. For point and space group prediction, balanced accuracies above 95% were obtained. The model demonstrated high performance for symmetry-distinct classes, including cubic crystal systems, point groups 3m and m-3m, and space groups Pnma and Pnnn. This work highlights the potential of ML for XRD-based structural characterization and accelerated discovery of perovskite materials
* 37 pages, 7 figures. Author accepted manuscript. Published in Engineering Applications of Artificial Intelligence
Quantum Kernel Machine Learning for Autonomous Materials Science
Jan 16, 2026Autonomous materials science, where active learning is used to navigate large compositional phase space, has emerged as a powerful vehicle to rapidly explore new materials. A crucial aspect of autonomous materials science is exploring new materials using as little data as possible. Gaussian process-based active learning allows effective charting of multi-dimensional parameter space with a limited number of training data, and thus is a common algorithmic choice for autonomous materials science. An integral part of the autonomous workflow is the application of kernel functions for quantifying similarities among measured data points. A recent theoretical breakthrough has shown that quantum kernel models can achieve similar performance with less training data than classical models. This signals the possible advantage of applying quantum kernel machine learning to autonomous materials discovery. In this work, we compare quantum and classical kernels for their utility in sequential phase space navigation for autonomous materials science. Specifically, we compute a quantum kernel and several classical kernels for x-ray diffraction patterns taken from an Fe-Ga-Pd ternary composition spread library. We conduct our study on both IonQ's Aria trapped ion quantum computer hardware and the corresponding classical noisy simulator. We experimentally verify that a quantum kernel model can outperform some classical kernel models. The results highlight the potential of quantum kernel machine learning methods for accelerating materials discovery and suggest complex x-ray diffraction data is a candidate for robust quantum kernel model advantage.
Revisiting the Broken Symmetry Phase of Solid Hydrogen: A Neural Network Variational Monte Carlo Study
Dec 19, 2025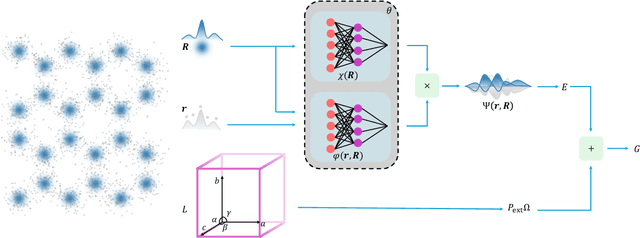
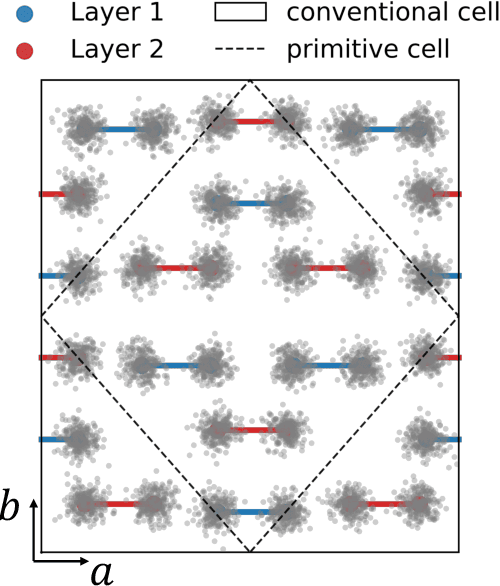
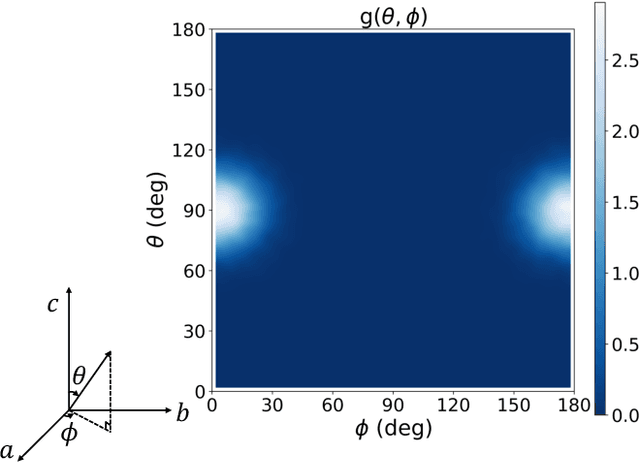
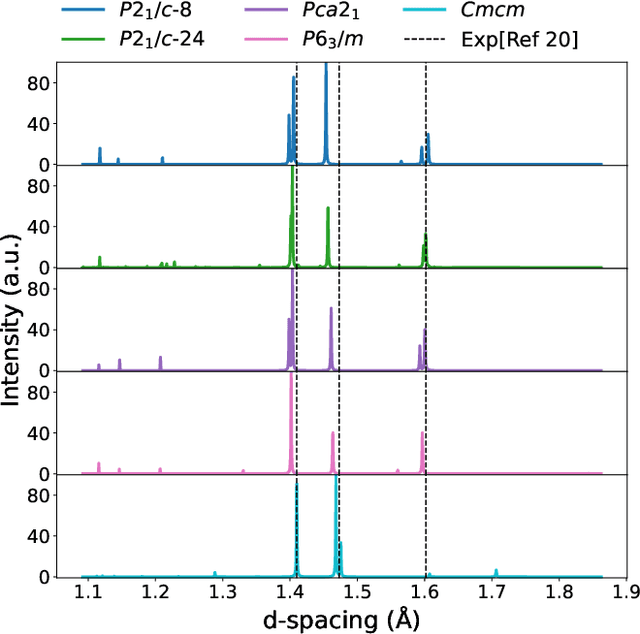
The crystal structure of high-pressure solid hydrogen remains a fundamental open problem. Although the research frontier has mostly shifted toward ultra-high pressure phases above 400 GPa, we show that even the broken symmetry phase observed around 130~GPa requires revisiting due to its intricate coupling of electronic and nuclear degrees of freedom. Here, we develop a first principle quantum Monte Carlo framework based on a deep neural network wave function that treats both electrons and nuclei quantum mechanically within the constant pressure ensemble. Our calculations reveal an unreported ground-state structure candidate for the broken symmetry phase with $Cmcm$ space group symmetry, and we test its stability up to 96 atoms. The predicted structure quantitatively matches the experimental equation of state and X-ray diffraction patterns. Furthermore, our group-theoretical analysis shows that the $Cmcm$ structure is compatible with existing Raman and infrared spectroscopic data. Crucially, static density functional theory calculation reveals the $Cmcm$ structure as a dynamically unstable saddle point on the Born-Oppenheimer potential energy surface, demonstrating that a full quantum many-body treatment of the problem is necessary. These results shed new light on the phase diagram of high-pressure hydrogen and call for further experimental verifications.
AGAPI-Agents: An Open-Access Agentic AI Platform for Accelerated Materials Design on AtomGPT.org
Dec 12, 2025Artificial intelligence is reshaping scientific discovery, yet its use in materials research remains limited by fragmented computational ecosystems, reproducibility challenges, and dependence on commercial large language models (LLMs). Here we introduce AGAPI (AtomGPT.org API), an open-access agentic AI platform that integrates more than eight open-source LLMs with over twenty materials-science API endpoints, unifying databases, simulation tools, and machine-learning models through a common orchestration framework. AGAPI employs an Agent-Planner-Executor-Summarizer architecture that autonomously constructs and executes multi-step workflows spanning materials data retrieval, graph neural network property prediction, machine-learning force-field optimization, tight-binding calculations, diffraction analysis, and inverse design. We demonstrate AGAPI through end-to-end workflows, including heterostructure construction, powder X-ray diffraction analysis, and semiconductor defect engineering requiring up to ten sequential operations. In addition, we evaluate AGAPI using 30+ example prompts as test cases and compare agentic predictions with and without tool access against experimental data. With more than 1,000 active users, AGAPI provides a scalable and transparent foundation for reproducible, AI-accelerated materials discovery. AGAPI-Agents codebase is available at https://github.com/atomgptlab/agapi.
Completion of partial structures using Patterson maps with the CrysFormer machine learning model
Nov 13, 2025



Protein structure determination has long been one of the primary challenges of structural biology, to which deep machine learning (ML)-based approaches have increasingly been applied. However, these ML models generally do not incorporate the experimental measurements directly, such as X-ray crystallographic diffraction data. To this end, we explore an approach that more tightly couples these traditional crystallographic and recent ML-based methods, by training a hybrid 3-d vision transformer and convolutional network on inputs from both domains. We make use of two distinct input constructs / Patterson maps, which are directly obtainable from crystallographic data, and ``partial structure'' template maps derived from predicted structures deposited in the AlphaFold Protein Structure Database with subsequently omitted residues. With these, we predict electron density maps that are then post-processed into atomic models through standard crystallographic refinement processes. Introducing an initial dataset of small protein fragments taken from Protein Data Bank entries and placing them in hypothetical crystal settings, we demonstrate that our method is effective at both improving the phases of the crystallographic structure factors and completing the regions missing from partial structure templates, as well as improving the agreement of the electron density maps with the ground truth atomic structures.
Learning neural representations for X-ray ptychography reconstruction with unknown probes
Sep 04, 2025



X-ray ptychography provides exceptional nanoscale resolution and is widely applied in materials science, biology, and nanotechnology. However, its full potential is constrained by the critical challenge of accurately reconstructing images when the illuminating probe is unknown. Conventional iterative methods and deep learning approaches are often suboptimal, particularly under the low-signal conditions inherent to low-dose and high-speed experiments. These limitations compromise reconstruction fidelity and restrict the broader adoption of the technique. In this work, we introduce the Ptychographic Implicit Neural Representation (PtyINR), a self-supervised framework that simultaneously addresses the object and probe recovery problem. By parameterizing both as continuous neural representations, PtyINR performs end-to-end reconstruction directly from raw diffraction patterns without requiring any pre-characterization of the probe. Extensive evaluations demonstrate that PtyINR achieves superior reconstruction quality on both simulated and experimental data, with remarkable robustness under challenging low-signal conditions. Furthermore, PtyINR offers a generalizable, physics-informed framework for addressing probe-dependent inverse problems, making it applicable to a wide range of computational microscopy problems.
Building-Block Aware Generative Modeling for 3D Crystals of Metal Organic Frameworks
May 13, 2025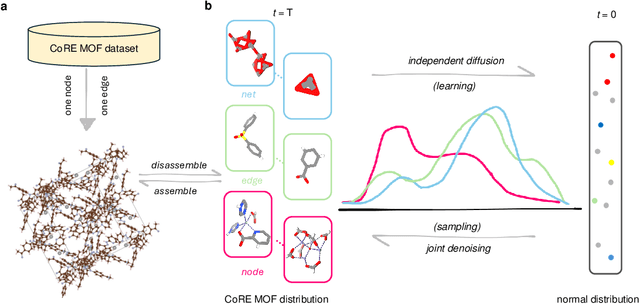
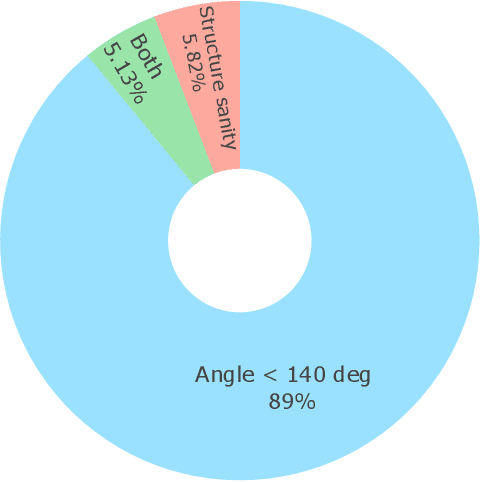
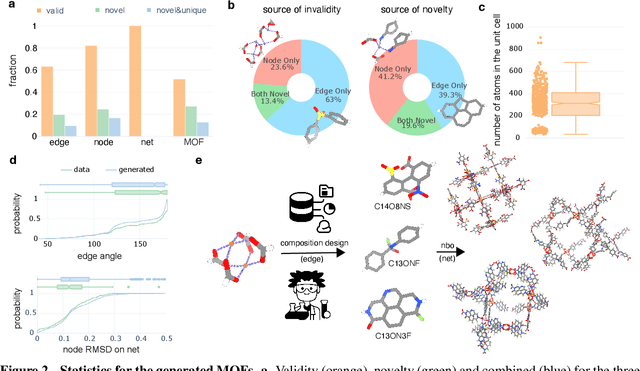
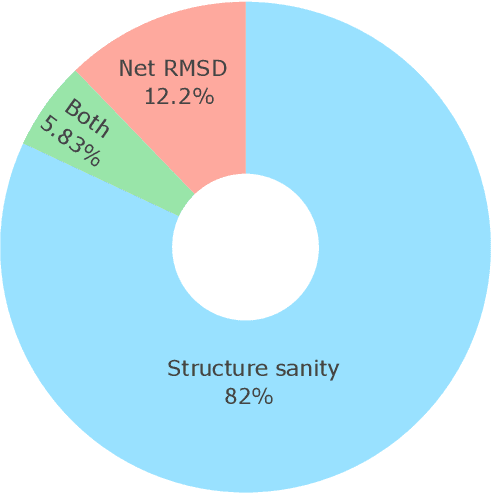
Metal-organic frameworks (MOFs) marry inorganic nodes, organic edges, and topological nets into programmable porous crystals, yet their astronomical design space defies brute-force synthesis. Generative modeling holds ultimate promise, but existing models either recycle known building blocks or are restricted to small unit cells. We introduce Building-Block-Aware MOF Diffusion (BBA MOF Diffusion), an SE(3)-equivariant diffusion model that learns 3D all-atom representations of individual building blocks, encoding crystallographic topological nets explicitly. Trained on the CoRE-MOF database, BBA MOF Diffusion readily samples MOFs with unit cells containing 1000 atoms with great geometric validity, novelty, and diversity mirroring experimental databases. Its native building-block representation produces unprecedented metal nodes and organic edges, expanding accessible chemical space by orders of magnitude. One high-scoring [Zn(1,4-TDC)(EtOH)2] MOF predicted by the model was synthesized, where powder X-ray diffraction, thermogravimetric analysis, and N2 sorption confirm its structural fidelity. BBA-Diff thus furnishes a practical pathway to synthesizable and high-performing MOFs.
opXRD: Open Experimental Powder X-ray Diffraction Database
Mar 07, 2025Powder X-ray diffraction (pXRD) experiments are a cornerstone for materials structure characterization. Despite their widespread application, analyzing pXRD diffractograms still presents a significant challenge to automation and a bottleneck in high-throughput discovery in self-driving labs. Machine learning promises to resolve this bottleneck by enabling automated powder diffraction analysis. A notable difficulty in applying machine learning to this domain is the lack of sufficiently sized experimental datasets, which has constrained researchers to train primarily on simulated data. However, models trained on simulated pXRD patterns showed limited generalization to experimental patterns, particularly for low-quality experimental patterns with high noise levels and elevated backgrounds. With the Open Experimental Powder X-Ray Diffraction Database (opXRD), we provide an openly available and easily accessible dataset of labeled and unlabeled experimental powder diffractograms. Labeled opXRD data can be used to evaluate the performance of models on experimental data and unlabeled opXRD data can help improve the performance of models on experimental data, e.g. through transfer learning methods. We collected \numpatterns diffractograms, 2179 of them labeled, from a wide spectrum of materials classes. We hope this ongoing effort can guide machine learning research toward fully automated analysis of pXRD data and thus enable future self-driving materials labs.
A Start To End Machine Learning Approach To Maximize Scientific Throughput From The LCLS-II-HE
May 29, 2025With the increasing brightness of Light sources, including the Diffraction-Limited brightness upgrade of APS and the high-repetition-rate upgrade of LCLS, the proposed experiments therein are becoming increasingly complex. For instance, experiments at LCLS-II-HE will require the X-ray beam to be within a fraction of a micron in diameter, with pointing stability of a few nanoradians, at the end of a kilometer-long electron accelerator, a hundred-meter-long undulator section, and tens of meters long X-ray optics. This enhancement of brightness will increase the data production rate to rival the largest data generators in the world. Without real-time active feedback control and an optimized pipeline to transform measurements to scientific information and insights, researchers will drown in a deluge of mostly useless data, and fail to extract the highly sophisticated insights that the recent brightness upgrades promise. In this article, we outline the strategy we are developing at SLAC to implement Machine Learning driven optimization, automation and real-time knowledge extraction from the electron-injector at the start of the electron accelerator, to the multidimensional X-ray optical systems, and till the experimental endstations and the high readout rate, multi-megapixel detectors at LCLS to deliver the design performance to the users. This is illustrated via examples from Accelerator, Optics and End User applications.
deCIFer: Crystal Structure Prediction from Powder Diffraction Data using Autoregressive Language Models
Feb 04, 2025Novel materials drive progress across applications from energy storage to electronics. Automated characterization of material structures with machine learning methods offers a promising strategy for accelerating this key step in material design. In this work, we introduce an autoregressive language model that performs crystal structure prediction (CSP) from powder diffraction data. The presented model, deCIFer, generates crystal structures in the widely used Crystallographic Information File (CIF) format and can be conditioned on powder X-ray diffraction (PXRD) data. Unlike earlier works that primarily rely on high-level descriptors like composition, deCIFer performs CSP from diffraction data. We train deCIFer on nearly 2.3M unique crystal structures and validate on diverse sets of PXRD patterns for characterizing challenging inorganic crystal systems. Qualitative and quantitative assessments using the residual weighted profile and Wasserstein distance show that deCIFer produces structures that more accurately match the target diffraction data when conditioned, compared to the unconditioned case. Notably, deCIFer can achieve a 94% match rate on unseen data. deCIFer bridges experimental diffraction data with computational CSP, lending itself as a powerful tool for crystal structure characterization and accelerating materials discovery.