 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBuilding-Block Aware Generative Modeling for 3D Crystals of Metal Organic Frameworks
May 13, 2025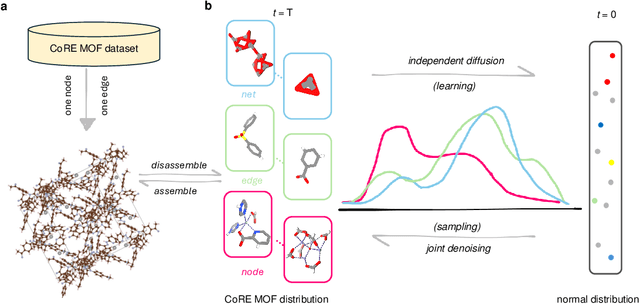
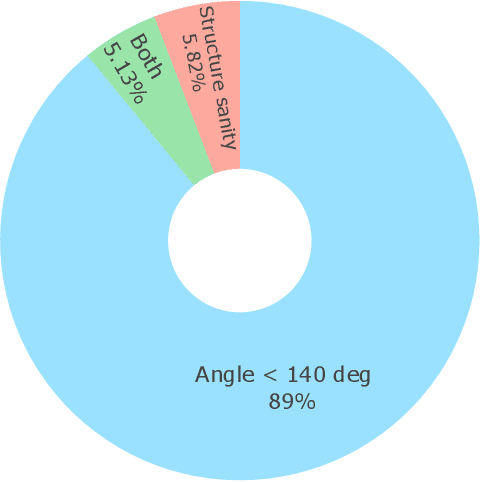
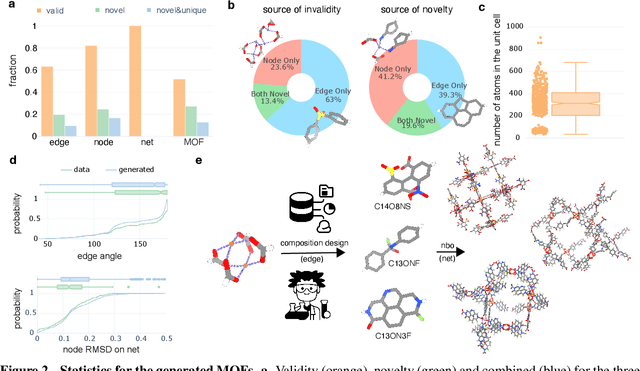
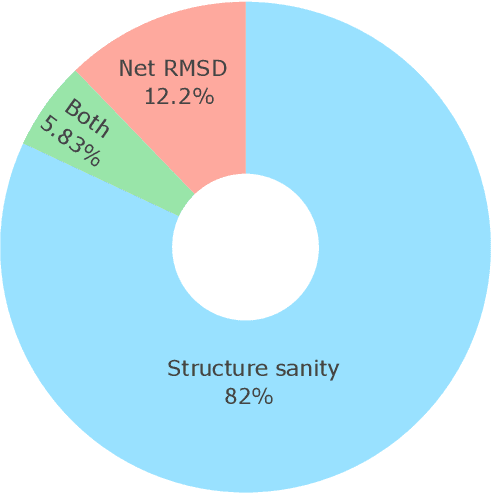
Metal-organic frameworks (MOFs) marry inorganic nodes, organic edges, and topological nets into programmable porous crystals, yet their astronomical design space defies brute-force synthesis. Generative modeling holds ultimate promise, but existing models either recycle known building blocks or are restricted to small unit cells. We introduce Building-Block-Aware MOF Diffusion (BBA MOF Diffusion), an SE(3)-equivariant diffusion model that learns 3D all-atom representations of individual building blocks, encoding crystallographic topological nets explicitly. Trained on the CoRE-MOF database, BBA MOF Diffusion readily samples MOFs with unit cells containing 1000 atoms with great geometric validity, novelty, and diversity mirroring experimental databases. Its native building-block representation produces unprecedented metal nodes and organic edges, expanding accessible chemical space by orders of magnitude. One high-scoring [Zn(1,4-TDC)(EtOH)2] MOF predicted by the model was synthesized, where powder X-ray diffraction, thermogravimetric analysis, and N2 sorption confirm its structural fidelity. BBA-Diff thus furnishes a practical pathway to synthesizable and high-performing MOFs.
Score-based generative diffusion with "active" correlated noise sources
Nov 11, 2024



Diffusion models exhibit robust generative properties by approximating the underlying distribution of a dataset and synthesizing data by sampling from the approximated distribution. In this work, we explore how the generative performance may be be modulated if noise sources with temporal correlations -- akin to those used in the field of active matter -- are used for the destruction of the data in the forward process. Our numerical and analytical experiments suggest that the corresponding reverse process may exhibit improved generative properties.
A Database of Ultrastable MOFs Reassembled from Stable Fragments with Machine Learning Models
Oct 25, 2022



High-throughput screening of large hypothetical databases of metal-organic frameworks (MOFs) can uncover new materials, but their stability in real-world applications is often unknown. We leverage community knowledge and machine learning (ML) models to identify MOFs that are thermally stable and stable upon activation. We separate these MOFs into their building blocks and recombine them to make a new hypothetical MOF database of over 50,000 structures that samples orders of magnitude more connectivity nets and inorganic building blocks than prior databases. This database shows an order of magnitude enrichment of ultrastable MOF structures that are stable upon activation and more than one standard deviation more thermally stable than the average experimentally characterized MOF. For the nearly 10,000 ultrastable MOFs, we compute bulk elastic moduli to confirm these materials have good mechanical stability, and we report methane deliverable capacities. Our work identifies privileged metal nodes in ultrastable MOFs that optimize gas storage and mechanical stability simultaneously.
Low-cost machine learning approach to the prediction of transition metal phosphor excited state properties
Sep 18, 2022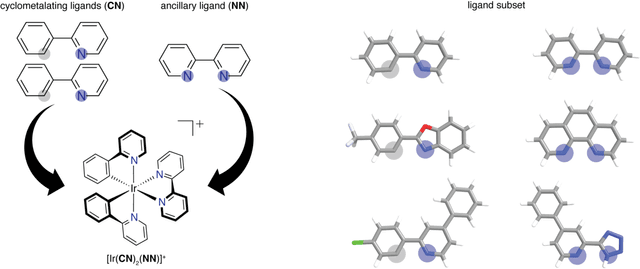
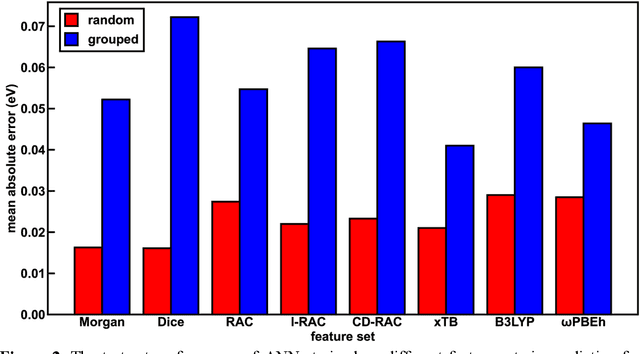
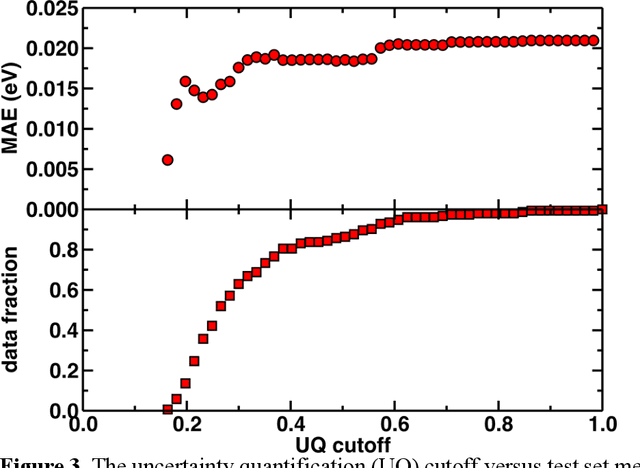
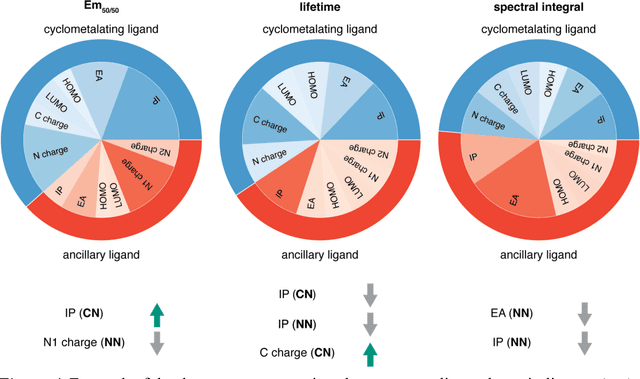
Photoactive iridium complexes are of broad interest due to their applications ranging from lighting to photocatalysis. However, the excited state property prediction of these complexes challenges ab initio methods such as time-dependent density functional theory (TDDFT) both from an accuracy and a computational cost perspective, complicating high throughput virtual screening (HTVS). We instead leverage low-cost machine learning (ML) models to predict the excited state properties of photoactive iridium complexes. We use experimental data of 1,380 iridium complexes to train and evaluate the ML models and identify the best-performing and most transferable models to be those trained on electronic structure features from low-cost density functional theory tight binding calculations. Using these models, we predict the three excited state properties considered, mean emission energy of phosphorescence, excited state lifetime, and emission spectral integral, with accuracy competitive with or superseding TDDFT. We conduct feature importance analysis to identify which iridium complex attributes govern excited state properties and we validate these trends with explicit examples. As a demonstration of how our ML models can be used for HTVS and the acceleration of chemical discovery, we curate a set of novel hypothetical iridium complexes and identify promising ligands for the design of new phosphors.
Rapid Exploration of a 32.5M Compound Chemical Space with Active Learning to Discover Density Functional Approximation Insensitive and Synthetically Accessible Transitional Metal Chromophores
Aug 10, 2022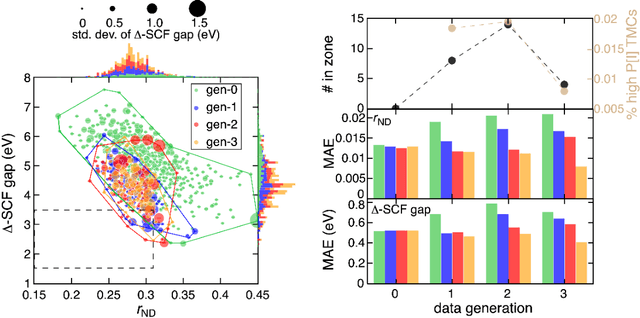
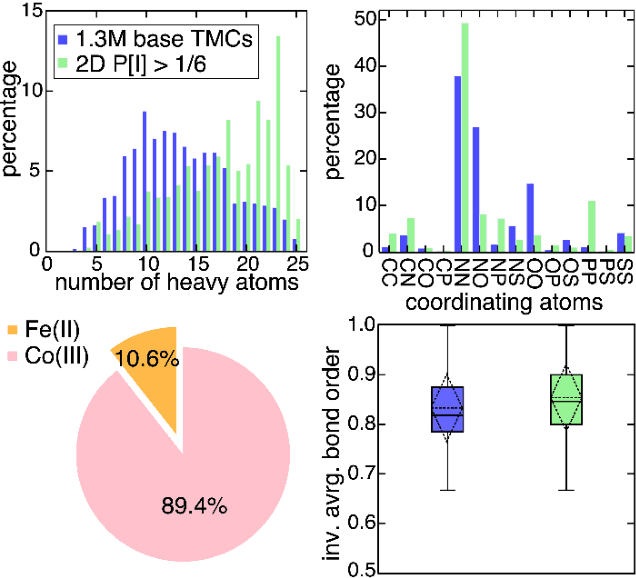
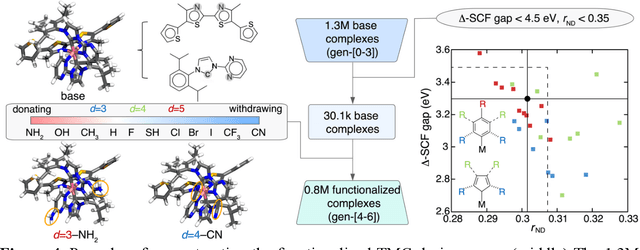
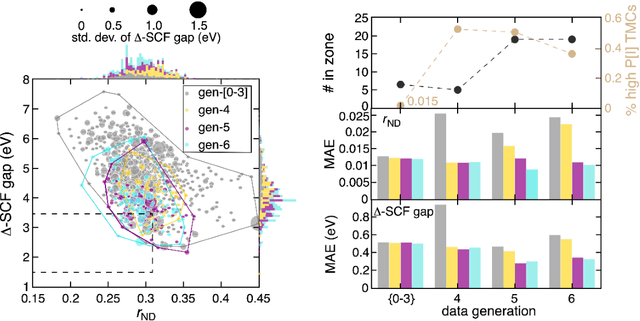
Two outstanding challenges for machine learning (ML) accelerated chemical discovery are the synthesizability of candidate molecules or materials and the fidelity of the data used in ML model training. To address the first challenge, we construct a hypothetical design space of 32.5M transition metal complexes (TMCs), in which all of the constituent fragments (i.e., metals and ligands) and ligand symmetries are synthetically accessible. To address the second challenge, we search for consensus in predictions among 23 density functional approximations across multiple rungs of Jacob's ladder. To accelerate the screening of these 32.5M TMCs, we use efficient global optimization to sample candidate low-spin chromophores that simultaneously have low absorption energies and low static correlation. Despite the scarcity (i.e., $<$ 0.01\%) of potential chromophores in this large chemical space, we identify transition metal chromophores with high likelihood (i.e., $>$ 10\%) as the ML models improve during active learning. This represents a 1,000 fold acceleration in discovery corresponding to discoveries in days instead of years. Analyses of candidate chromophores reveal a preference for Co(III) and large, strong-field ligands with more bond saturation. We compute the absorption spectra of promising chromophores on the Pareto front by time-dependent density functional theory calculations and verify that two thirds of them have desired excited state properties. Although these complexes have never been experimentally explored, their constituent ligands demonstrated interesting optical properties in literature, exemplifying the effectiveness of our construction of realistic TMC design space and active learning approach.
A Transferable Recommender Approach for Selecting the Best Density Functional Approximations in Chemical Discovery
Jul 21, 2022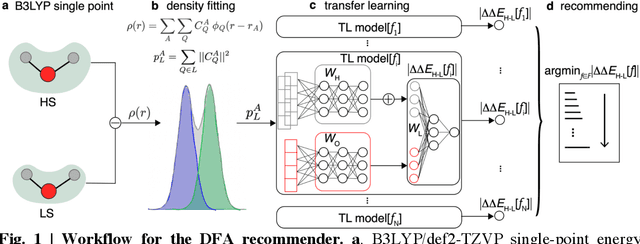
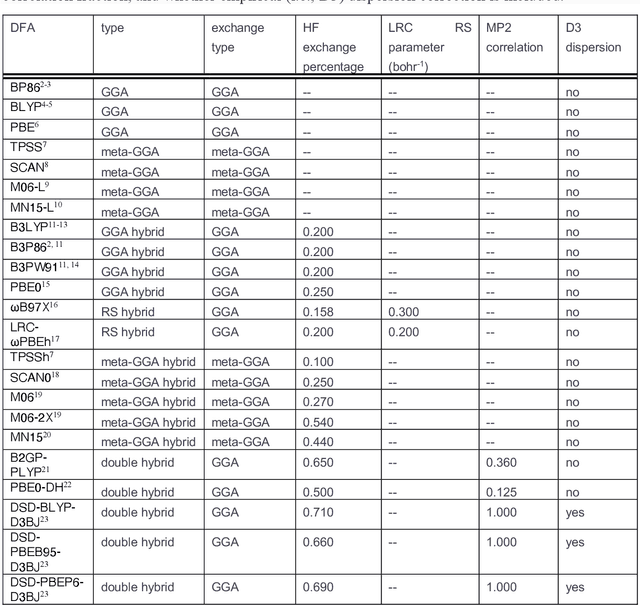
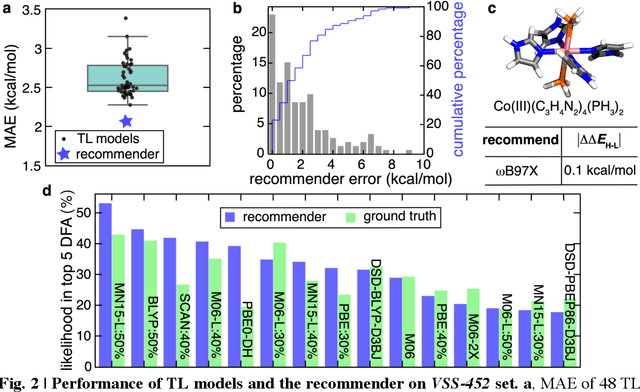
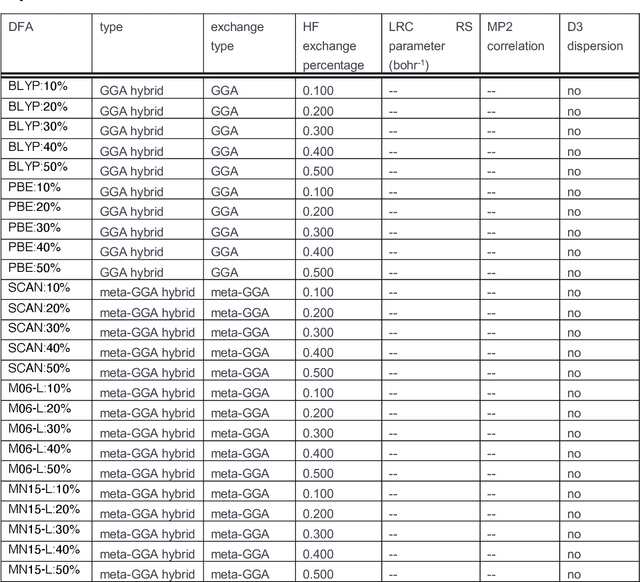
Approximate density functional theory (DFT) has become indispensable owing to its cost-accuracy trade-off in comparison to more computationally demanding but accurate correlated wavefunction theory. To date, however, no single density functional approximation (DFA) with universal accuracy has been identified, leading to uncertainty in the quality of data generated from DFT. With electron density fitting and transfer learning, we build a DFA recommender that selects the DFA with the lowest expected error with respect to gold standard but cost-prohibitive coupled cluster theory in a system-specific manner. We demonstrate this recommender approach on vertical spin-splitting energy evaluation for challenging transition metal complexes. Our recommender predicts top-performing DFAs and yields excellent accuracy (ca. 2 kcal/mol) for chemical discovery, outperforming both individual transfer learning models and the single best functional in a set of 48 DFAs. We demonstrate the transferability of the DFA recommender to experimentally synthesized compounds with distinct chemistry.
Putting Density Functional Theory to the Test in Machine-Learning-Accelerated Materials Discovery
May 06, 2022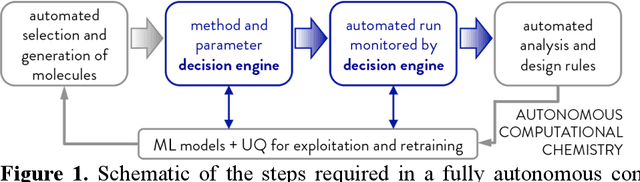
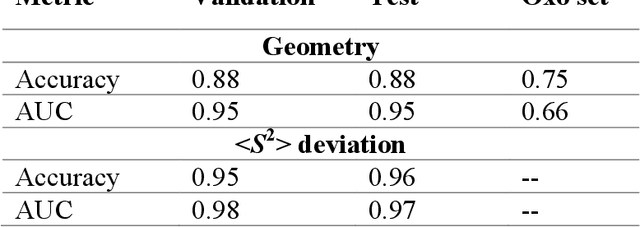
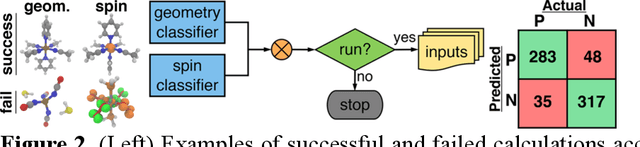
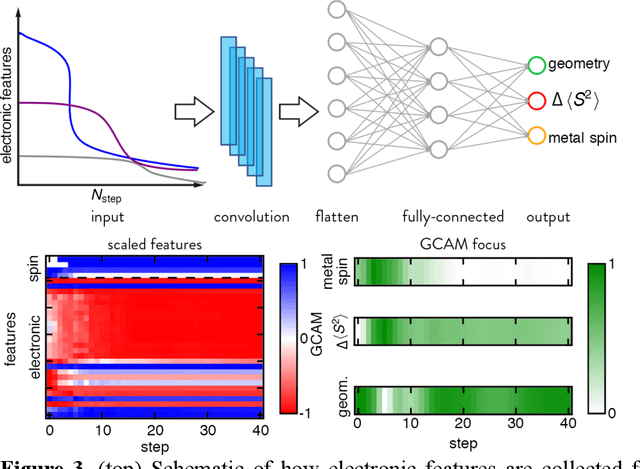
Accelerated discovery with machine learning (ML) has begun to provide the advances in efficiency needed to overcome the combinatorial challenge of computational materials design. Nevertheless, ML-accelerated discovery both inherits the biases of training data derived from density functional theory (DFT) and leads to many attempted calculations that are doomed to fail. Many compelling functional materials and catalytic processes involve strained chemical bonds, open-shell radicals and diradicals, or metal-organic bonds to open-shell transition-metal centers. Although promising targets, these materials present unique challenges for electronic structure methods and combinatorial challenges for their discovery. In this Perspective, we describe the advances needed in accuracy, efficiency, and approach beyond what is typical in conventional DFT-based ML workflows. These challenges have begun to be addressed through ML models trained to predict the results of multiple methods or the differences between them, enabling quantitative sensitivity analysis. For DFT to be trusted for a given data point in a high-throughput screen, it must pass a series of tests. ML models that predict the likelihood of calculation success and detect the presence of strong correlation will enable rapid diagnoses and adaptation strategies. These "decision engines" represent the first steps toward autonomous workflows that avoid the need for expert determination of the robustness of DFT-based materials discoveries.
Machine learning models predict calculation outcomes with the transferability necessary for computational catalysis
Mar 02, 2022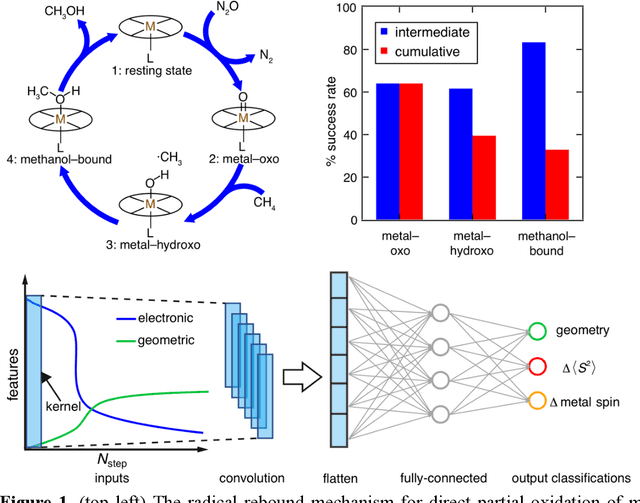
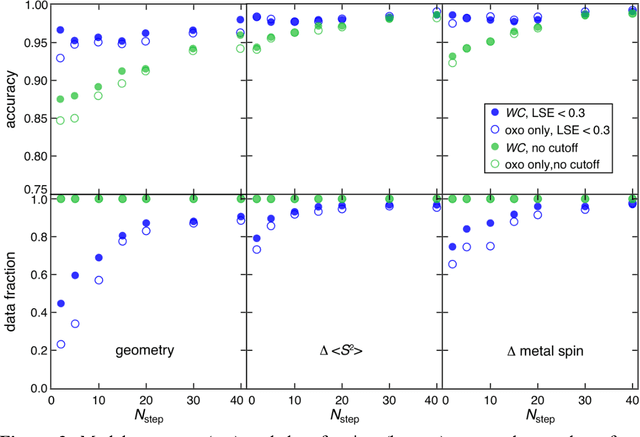
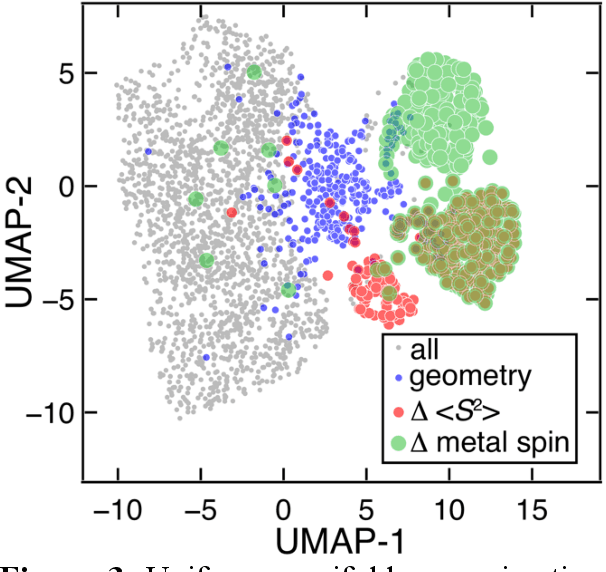
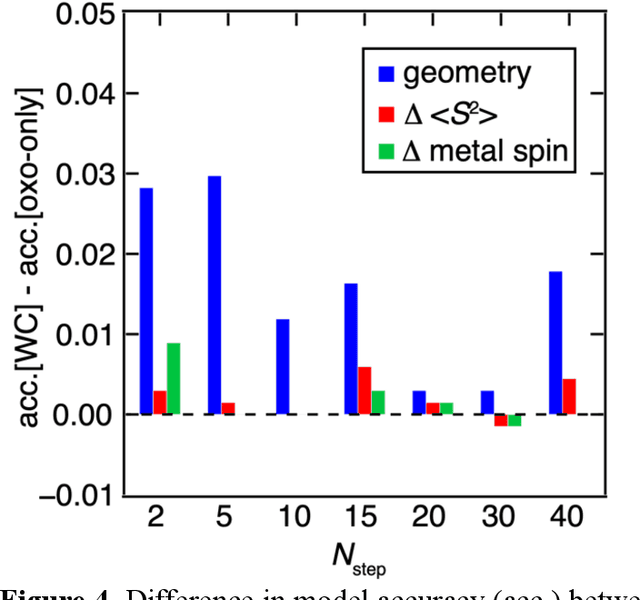
Virtual high throughput screening (VHTS) and machine learning (ML) have greatly accelerated the design of single-site transition-metal catalysts. VHTS of catalysts, however, is often accompanied with high calculation failure rate and wasted computational resources due to the difficulty of simultaneously converging all mechanistically relevant reactive intermediates to expected geometries and electronic states. We demonstrate a dynamic classifier approach, i.e., a convolutional neural network that monitors geometry optimization on the fly, and exploit its good performance and transferability for catalyst design. We show that the dynamic classifier performs well on all reactive intermediates in the representative catalytic cycle of the radical rebound mechanism for methane-to-methanol despite being trained on only one reactive intermediate. The dynamic classifier also generalizes to chemically distinct intermediates and metal centers absent from the training data without loss of accuracy or model confidence. We rationalize this superior model transferability to the use of on-the-fly electronic structure and geometric information generated from density functional theory calculations and the convolutional layer in the dynamic classifier. Combined with model uncertainty quantification, the dynamic classifier saves more than half of the computational resources that would have been wasted on unsuccessful calculations for all reactive intermediates being considered.
Two Wrongs Can Make a Right: A Transfer Learning Approach for Chemical Discovery with Chemical Accuracy
Jan 11, 2022Appropriately identifying and treating molecules and materials with significant multi-reference (MR) character is crucial for achieving high data fidelity in virtual high throughput screening (VHTS). Nevertheless, most VHTS is carried out with approximate density functional theory (DFT) using a single functional. Despite development of numerous MR diagnostics, the extent to which a single value of such a diagnostic indicates MR effect on chemical property prediction is not well established. We evaluate MR diagnostics of over 10,000 transition metal complexes (TMCs) and compare to those in organic molecules. We reveal that only some MR diagnostics are transferable across these materials spaces. By studying the influence of MR character on chemical properties (i.e., MR effect) that involves multiple potential energy surfaces (i.e., adiabatic spin splitting, $\Delta E_\mathrm{H-L}$, and ionization potential, IP), we observe that cancellation in MR effect outweighs accumulation. Differences in MR character are more important than the total degree of MR character in predicting MR effect in property prediction. Motivated by this observation, we build transfer learning models to directly predict CCSD(T)-level adiabatic $\Delta E_\mathrm{H-L}$ and IP from lower levels of theory. By combining these models with uncertainty quantification and multi-level modeling, we introduce a multi-pronged strategy that accelerates data acquisition by at least a factor of three while achieving chemical accuracy (i.e., 1 kcal/mol) for robust VHTS.
Audacity of huge: overcoming challenges of data scarcity and data quality for machine learning in computational materials discovery
Nov 02, 2021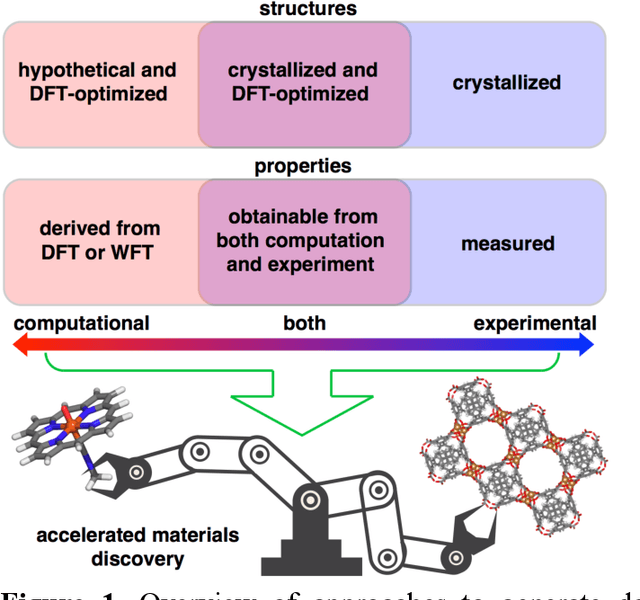
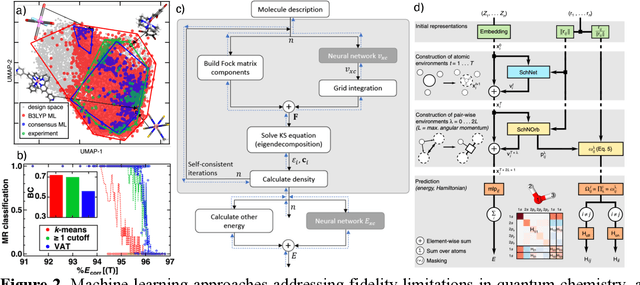
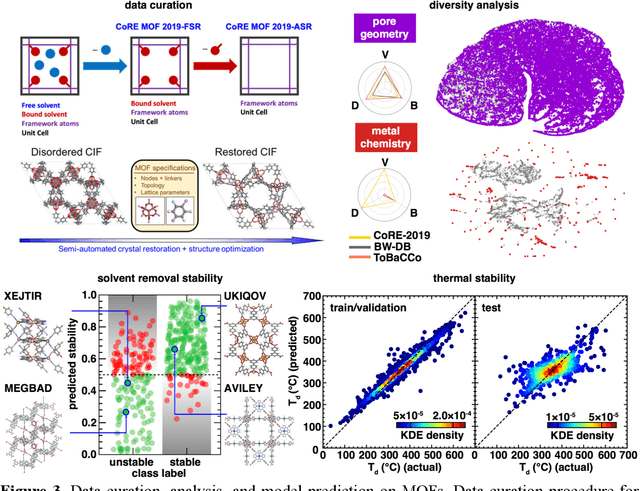
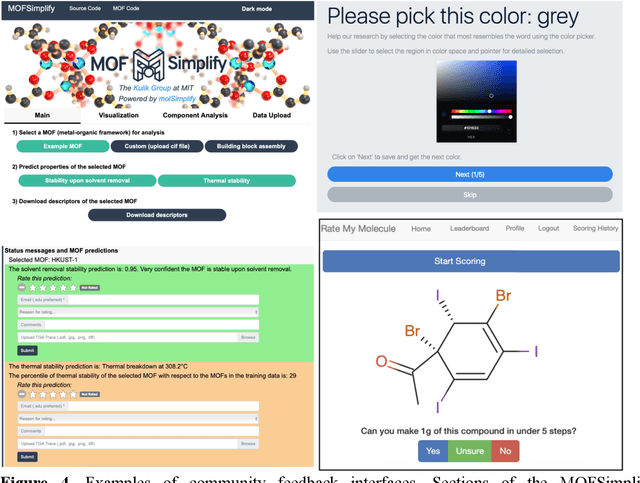
Machine learning (ML)-accelerated discovery requires large amounts of high-fidelity data to reveal predictive structure-property relationships. For many properties of interest in materials discovery, the challenging nature and high cost of data generation has resulted in a data landscape that is both scarcely populated and of dubious quality. Data-driven techniques starting to overcome these limitations include the use of consensus across functionals in density functional theory, the development of new functionals or accelerated electronic structure theories, and the detection of where computationally demanding methods are most necessary. When properties cannot be reliably simulated, large experimental data sets can be used to train ML models. In the absence of manual curation, increasingly sophisticated natural language processing and automated image analysis are making it possible to learn structure-property relationships from the literature. Models trained on these data sets will improve as they incorporate community feedback.