 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeM$^3$Exam: Benchmarking Multimodal Memory for Realistic User-Agent Interactions
Jun 05, 2026Language agents are increasingly deployed over accumulating multimodal information, yet existing benchmarks assume a human-human form with sparse visuals and straightforward content, evaluating neither reasoning over authentic multimodal file interaction nor the interpretation of concealed user information. We therefore introduce M$^3$Exam, a query-centric multimodal conversational memory benchmark built on realistic user-agent interaction, with multi-dimensional evaluation spanning cross-modal grounding and implicit information inference. Benchmarking MLLMs and memory systems reveals persistent gaps in cross-modal grounding, cross session reasoning, and the efficiency cost of accumulating multimodal context. We further propose M$^3$Proctor, a multimodal memory method that detects query modality bias and consumes raw visual sources only on demand, improving accuracy by 13% while cutting index-construction time and retrieved tokens by over 70%.
LifeSide: Benchmarking Agents as Lifelong Digital Companions
Jun 03, 2026Lifelong digital companions must integrate cross-session cues, continually update their understanding of users, and adapt to shifting privacy boundaries. Existing evaluations fail to capture this, testing memory recall and short-term empathy in isolation. To bridge this gap, we introduce \benchmark, a benchmark centered on multi-session \textit{Memory-Emotion-Environment} loops. By modeling users as persistent worlds with layered profiles and event trajectories, \benchmark uses multi-agent simulation to project environmental dynamics into dialogue, preserving the critical gap between latent thoughts and observable expressions. Evaluating 2,000 personas and 111K tasks across memory tracking, user understanding, privacy control, and emotional companionship, our experiment results reveal a stark reality: even models that saturate current memory benchmarks fail to sustain accurate user understanding and true companionship over long horizons.
Back to Basics: Let Conversational Agents Remember with Just Retrieval and Generation
Apr 13, 2026Existing conversational memory systems rely on complex hierarchical summarization or reinforcement learning to manage long-term dialogue history, yet remain vulnerable to context dilution as conversations grow. In this work, we offer a different perspective: the primary bottleneck may lie not in memory architecture, but in the \textit{Signal Sparsity Effect} within the latent knowledge manifold. Through controlled experiments, we identify two key phenomena: \textit{Decisive Evidence Sparsity}, where relevant signals become increasingly isolated with longer sessions, leading to sharp degradation in aggregation-based methods; and \textit{Dual-Level Redundancy}, where both inter-session interference and intra-session conversational filler introduce large amounts of non-informative content, hindering effective generation. Motivated by these insights, we propose \method, a minimalist framework that brings conversational memory back to basics, relying solely on retrieval and generation via Turn Isolation Retrieval (TIR) and Query-Driven Pruning (QDP). TIR replaces global aggregation with a max-activation strategy to capture turn-level signals, while QDP removes redundant sessions and conversational filler to construct a compact, high-density evidence set. Extensive experiments on multiple benchmarks demonstrate that \method achieves robust performance across diverse settings, consistently outperforming strong baselines while maintaining high efficiency in tokens and latency, establishing a new minimalist baseline for conversational memory.
UrbanFM: Scaling Urban Spatio-Temporal Foundation Models
Feb 24, 2026Urban systems, as dynamic complex systems, continuously generate spatio-temporal data streams that encode the fundamental laws of human mobility and city evolution. While AI for Science has witnessed the transformative power of foundation models in disciplines like genomics and meteorology, urban computing remains fragmented due to "scenario-specific" models, which are overfitted to specific regions or tasks, hindering their generalizability. To bridge this gap and advance spatio-temporal foundation models for urban systems, we adopt scaling as the central perspective and systematically investigate two key questions: what to scale and how to scale. Grounded in first-principles analysis, we identify three critical dimensions: heterogeneity, correlation, and dynamics, aligning these principles with the fundamental scientific properties of urban spatio-temporal data. Specifically, to address heterogeneity through data scaling, we construct WorldST. This billion-scale corpus standardizes diverse physical signals, such as traffic flow and speed, from over 100 global cities into a unified data format. To enable computation scaling for modeling correlations, we introduce the MiniST unit, a novel split mechanism that discretizes continuous spatio-temporal fields into learnable computational units to unify representations of grid-based and sensor-based observations. Finally, addressing dynamics via architecture scaling, we propose UrbanFM, a minimalist self-attention architecture designed with limited inductive biases to autonomously learn dynamic spatio-temporal dependencies from massive data. Furthermore, we establish EvalST, the largest-scale urban spatio-temporal benchmark to date. Extensive experiments demonstrate that UrbanFM achieves remarkable zero-shot generalization across unseen cities and tasks, marking a pivotal first step toward large-scale urban spatio-temporal foundation models.
Learning from Complexity: Exploring Dynamic Sample Pruning of Spatio-Temporal Training
Feb 22, 2026Spatio-temporal forecasting is fundamental to intelligent systems in transportation, climate science, and urban planning. However, training deep learning models on the massive, often redundant, datasets from these domains presents a significant computational bottleneck. Existing solutions typically focus on optimizing model architectures or optimizers, while overlooking the inherent inefficiency of the training data itself. This conventional approach of iterating over the entire static dataset each epoch wastes considerable resources on easy-to-learn or repetitive samples. In this paper, we explore a novel training-efficiency techniques, namely learning from complexity with dynamic sample pruning, ST-Prune, for spatio-temporal forecasting. Through dynamic sample pruning, we aim to intelligently identify the most informative samples based on the model's real-time learning state, thereby accelerating convergence and improving training efficiency. Extensive experiments conducted on real-world spatio-temporal datasets show that ST-Prune significantly accelerates the training speed while maintaining or even improving the model performance, and it also has scalability and universality.
Rationale-Grounded In-Context Learning for Time Series Reasoning with Multimodal Large Language Models
Jan 06, 2026The underperformance of existing multimodal large language models for time series reasoning lies in the absence of rationale priors that connect temporal observations to their downstream outcomes, which leads models to rely on superficial pattern matching rather than principled reasoning. We therefore propose the rationale-grounded in-context learning for time series reasoning, where rationales work as guiding reasoning units rather than post-hoc explanations, and develop the RationaleTS method. Specifically, we firstly induce label-conditioned rationales, composed of reasoning paths from observable evidence to the potential outcomes. Then, we design the hybrid retrieval by balancing temporal patterns and semantic contexts to retrieve correlated rationale priors for the final in-context inference on new samples. We conduct extensive experiments to demonstrate the effectiveness and efficiency of our proposed RationaleTS on three-domain time series reasoning tasks. We will release our code for reproduction.
Co-Learning: Code Learning for Multi-Agent Reinforcement Collaborative Framework with Conversational Natural Language Interfaces
Sep 02, 2024



Online question-and-answer (Q\&A) systems based on the Large Language Model (LLM) have progressively diverged from recreational to professional use. This paper proposed a Multi-Agent framework with environmentally reinforcement learning (E-RL) for code correction called Code Learning (Co-Learning) community, assisting beginners to correct code errors independently. It evaluates the performance of multiple LLMs from an original dataset with 702 error codes, uses it as a reward or punishment criterion for E-RL; Analyzes input error codes by the current agent; selects the appropriate LLM-based agent to achieve optimal error correction accuracy and reduce correction time. Experiment results showed that 3\% improvement in Precision score and 15\% improvement in time cost as compared with no E-RL method respectively. Our source code is available at: https://github.com/yuqian2003/Co_Learning
Deep Feature Embedding for Tabular Data
Aug 30, 2024



Tabular data learning has extensive applications in deep learning but its existing embedding techniques are limited in numerical and categorical features such as the inability to capture complex relationships and engineering. This paper proposes a novel deep embedding framework with leverages lightweight deep neural networks to generate effective feature embeddings for tabular data in machine learning research. For numerical features, a two-step feature expansion and deep transformation technique is used to capture copious semantic information. For categorical features, a unique identification vector for each entity is referred by a compact lookup table with a parameterized deep embedding function to uniform the embedding size dimensions, and transformed into a embedding vector using deep neural network. Experiments are conducted on real-world datasets for performance evaluation.
AMPCliff: quantitative definition and benchmarking of activity cliffs in antimicrobial peptides
Apr 15, 2024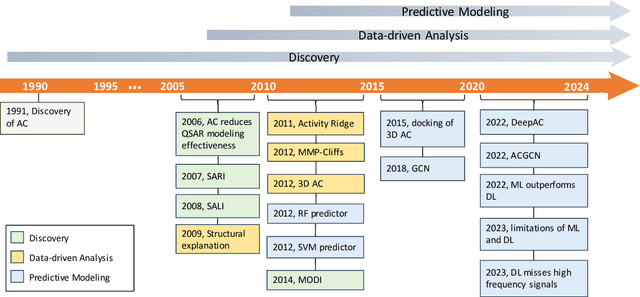

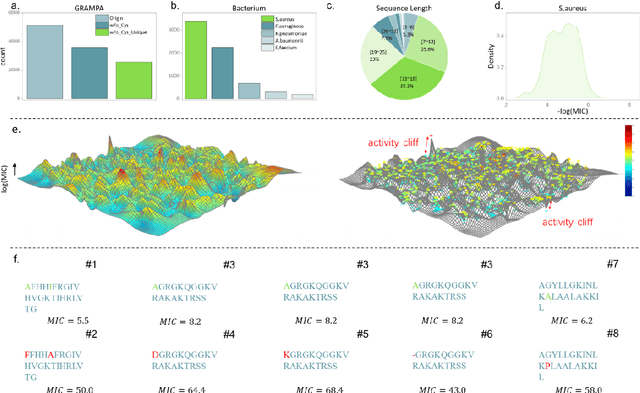
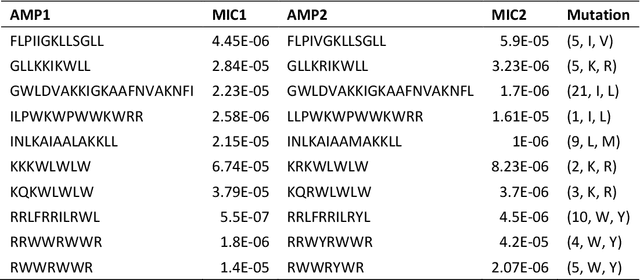
Activity cliff (AC) is a phenomenon that a pair of similar molecules differ by a small structural alternation but exhibit a large difference in their biochemical activities. The AC of small molecules has been extensively investigated but limited knowledge is accumulated about the AC phenomenon in peptides with canonical amino acids. This study introduces a quantitative definition and benchmarking framework AMPCliff for the AC phenomenon in antimicrobial peptides (AMPs) composed by canonical amino acids. A comprehensive analysis of the existing AMP dataset reveals a significant prevalence of AC within AMPs. AMPCliff quantifies the activities of AMPs by the metric minimum inhibitory concentration (MIC), and defines 0.9 as the minimum threshold for the normalized BLOSUM62 similarity score between a pair of aligned peptides with at least two-fold MIC changes. This study establishes a benchmark dataset of paired AMPs in Staphylococcus aureus from the publicly available AMP dataset GRAMPA, and conducts a rigorous procedure to evaluate various AMP AC prediction models, including nine machine learning, four deep learning algorithms, four masked language models, and four generative language models. Our analysis reveals that these models are capable of detecting AMP AC events and the pre-trained protein language ESM2 model demonstrates superior performance across the evaluations. The predictive performance of AMP activity cliffs remains to be further improved, considering that ESM2 with 33 layers only achieves the Spearman correlation coefficient=0.50 for the regression task of the MIC values on the benchmark dataset. Source code and additional resources are available at https://www.healthinformaticslab.org/supp/ or https://github.com/Kewei2023/AMPCliff-generation.
PepHarmony: A Multi-View Contrastive Learning Framework for Integrated Sequence and Structure-Based Peptide Encoding
Jan 21, 2024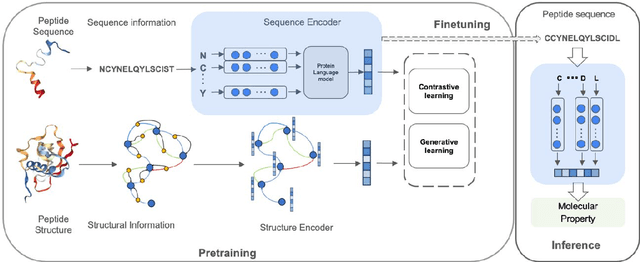
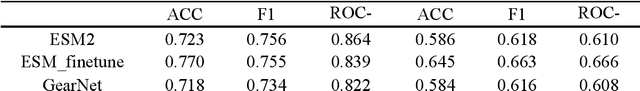
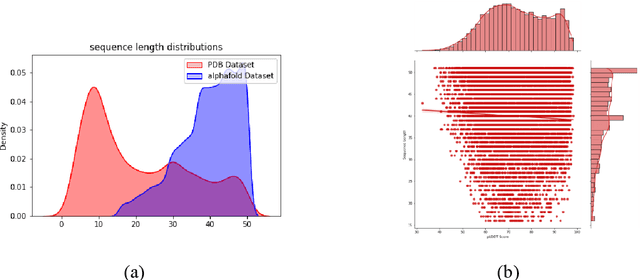

Recent advances in protein language models have catalyzed significant progress in peptide sequence representation. Despite extensive exploration in this field, pre-trained models tailored for peptide-specific needs remain largely unaddressed due to the difficulty in capturing the complex and sometimes unstable structures of peptides. This study introduces a novel multi-view contrastive learning framework PepHarmony for the sequence-based peptide encoding task. PepHarmony innovatively combines both sequence- and structure-level information into a sequence-level encoding module through contrastive learning. We carefully select datasets from the Protein Data Bank (PDB) and AlphaFold database to encompass a broad spectrum of peptide sequences and structures. The experimental data highlights PepHarmony's exceptional capability in capturing the intricate relationship between peptide sequences and structures compared with the baseline and fine-tuned models. The robustness of our model is confirmed through extensive ablation studies, which emphasize the crucial roles of contrastive loss and strategic data sorting in enhancing predictive performance. The proposed PepHarmony framework serves as a notable contribution to peptide representations, and offers valuable insights for future applications in peptide drug discovery and peptide engineering. We have made all the source code utilized in this study publicly accessible via GitHub at https://github.com/zhangruochi/PepHarmony or http://www.healthinformaticslab.org/supp/.