 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRetroAgent: Harnessing LLMs to Search Over Structured Memory for Agentic Retrosynthesis Planning
Jul 16, 2026Multi-step retrosynthesis planning seeks to decompose a target molecule into commercially available building blocks through a sequence of feasible reactions. The vast combinatorial search space makes this task challenging even for expert chemists. Traditional methods combine tree search with offline-trained value networks that score candidates in isolation, without reasoning about complete multi-step routes. Recent work leverages Large Language Models (LLMs) for this task, but relies on simple interfaces that limit exploration of the full search space. We introduce RetroAgent, an LLM agent that bridges symbolic search and neural reasoning through a harness with structured memory. Through memory and chemistry tools, the agent observes the full search state, including explored routes, available alternatives, and properties of intermediates, enabling informed decisions grounded in both global progress and domain knowledge. Experiments on in-distribution and out-of-distribution benchmarks demonstrate that RetroAgent delivers strong performance and generalization.
NeuroSonic: Conditional Flow Matching for EEG-to-Speech Reconstruction
Jun 23, 2026Reconstructing continuous speech from scalp electroencephalography (EEG) remains fundamentally challenging. EEG provides a weak, spatially diffuse, and highly variable measurement of distributed cortical activity, whereas speech is organized as a coherent acoustic trajectory with strong harmonic and temporal structure. The resulting mismatch makes waveform regression unstable and causes stochastic multi-step generation to be sensitive to artifact-dependent conditioning and subject variability. We introduce NeuroSonic, a conditional flow-matching framework for EEG-to-speech reconstruction. Instead of predicting waveforms directly or refining them through stochastic denoising, NeuroSonic learns a deterministic probability-flow velocity field that transports a noise-corrupted acoustic state toward clean speech under EEG conditioning. EEG and audio are embedded into a shared token space and processed by a time-conditioned gated Transformer that parameterizes the transport ordinary differential equation. This formulation models trajectory evolution explicitly while avoiding iterative stochastic sampling. We evaluate NeuroSonic on the CineBrain and EAV benchmarks under cross-subject evaluation. Across both datasets, the proposed method improves distributional realism, spectral fidelity, and perceptual quality over representative GAN-, diffusion-, and mean-flow baselines, with up to a 26.3\% gain in overall perceptual quality. The performance gap is most evident in artifact-heavy segments, where conditioning variability is strongest. These findings indicate that deterministic conditional transport provides a stable and effective formulation for EEG-driven speech reconstruction. Code is available at https://github.com/Y-Research-SBU/NeuroSonic/ .
Double-Ended Synthesis Planning with Goal-Constrained Bidirectional Search
Jul 08, 2024



Computer-aided synthesis planning (CASP) algorithms have demonstrated expert-level abilities in planning retrosynthetic routes to molecules of low to moderate complexity. However, current search methods assume the sufficiency of reaching arbitrary building blocks, failing to address the common real-world constraint where using specific molecules is desired. To this end, we present a formulation of synthesis planning with starting material constraints. Under this formulation, we propose Double-Ended Synthesis Planning (DESP), a novel CASP algorithm under a bidirectional graph search scheme that interleaves expansions from the target and from the goal starting materials to ensure constraint satisfiability. The search algorithm is guided by a goal-conditioned cost network learned offline from a partially observed hypergraph of valid chemical reactions. We demonstrate the utility of DESP in improving solve rates and reducing the number of search expansions by biasing synthesis planning towards expert goals on multiple new benchmarks. DESP can make use of existing one-step retrosynthesis models, and we anticipate its performance to scale as these one-step model capabilities improve.
Efficient Evolutionary Search Over Chemical Space with Large Language Models
Jun 23, 2024



Molecular discovery, when formulated as an optimization problem, presents significant computational challenges because optimization objectives can be non-differentiable. Evolutionary Algorithms (EAs), often used to optimize black-box objectives in molecular discovery, traverse chemical space by performing random mutations and crossovers, leading to a large number of expensive objective evaluations. In this work, we ameliorate this shortcoming by incorporating chemistry-aware Large Language Models (LLMs) into EAs. Namely, we redesign crossover and mutation operations in EAs using LLMs trained on large corpora of chemical information. We perform extensive empirical studies on both commercial and open-source models on multiple tasks involving property optimization, molecular rediscovery, and structure-based drug design, demonstrating that the joint usage of LLMs with EAs yields superior performance over all baseline models across single- and multi-objective settings. We demonstrate that our algorithm improves both the quality of the final solution and convergence speed, thereby reducing the number of required objective evaluations. Our code is available at http://github.com/zoom-wang112358/MOLLEO
AUTODIFF: Autoregressive Diffusion Modeling for Structure-based Drug Design
Apr 03, 2024Structure-based drug design (SBDD), which aims to generate molecules that can bind tightly to the target protein, is an essential problem in drug discovery, and previous approaches have achieved initial success. However, most existing methods still suffer from invalid local structure or unrealistic conformation issues, which are mainly due to the poor leaning of bond angles or torsional angles. To alleviate these problems, we propose AUTODIFF, a diffusion-based fragment-wise autoregressive generation model. Specifically, we design a novel molecule assembly strategy named conformal motif that preserves the conformation of local structures of molecules first, then we encode the interaction of the protein-ligand complex with an SE(3)-equivariant convolutional network and generate molecules motif-by-motif with diffusion modeling. In addition, we also improve the evaluation framework of SBDD by constraining the molecular weights of the generated molecules in the same range, together with some new metrics, which make the evaluation more fair and practical. Extensive experiments on CrossDocked2020 demonstrate that our approach outperforms the existing models in generating realistic molecules with valid structures and conformations while maintaining high binding affinity.
AS-FIBA: Adaptive Selective Frequency-Injection for Backdoor Attack on Deep Face Restoration
Mar 11, 2024



Deep learning-based face restoration models, increasingly prevalent in smart devices, have become targets for sophisticated backdoor attacks. These attacks, through subtle trigger injection into input face images, can lead to unexpected restoration outcomes. Unlike conventional methods focused on classification tasks, our approach introduces a unique degradation objective tailored for attacking restoration models. Moreover, we propose the Adaptive Selective Frequency Injection Backdoor Attack (AS-FIBA) framework, employing a neural network for input-specific trigger generation in the frequency domain, seamlessly blending triggers with benign images. This results in imperceptible yet effective attacks, guiding restoration predictions towards subtly degraded outputs rather than conspicuous targets. Extensive experiments demonstrate the efficacy of the degradation objective on state-of-the-art face restoration models. Additionally, it is notable that AS-FIBA can insert effective backdoors that are more imperceptible than existing backdoor attack methods, including WaNet, ISSBA, and FIBA.
Substrate Scope Contrastive Learning: Repurposing Human Bias to Learn Atomic Representations
Feb 19, 2024



Learning molecular representation is a critical step in molecular machine learning that significantly influences modeling success, particularly in data-scarce situations. The concept of broadly pre-training neural networks has advanced fields such as computer vision, natural language processing, and protein engineering. However, similar approaches for small organic molecules have not achieved comparable success. In this work, we introduce a novel pre-training strategy, substrate scope contrastive learning, which learns atomic representations tailored to chemical reactivity. This method considers the grouping of substrates and their yields in published substrate scope tables as a measure of their similarity or dissimilarity in terms of chemical reactivity. We focus on 20,798 aryl halides in the CAS Content Collection spanning thousands of publications to learn a representation of aryl halide reactivity. We validate our pre-training approach through both intuitive visualizations and comparisons to traditional reactivity descriptors and physical organic chemistry principles. The versatility of these embeddings is further evidenced in their application to yield prediction, regioselectivity prediction, and the diverse selection of new substrates. This work not only presents a chemistry-tailored neural network pre-training strategy to learn reactivity-aligned atomic representations, but also marks a first-of-its-kind approach to benefit from the human bias in substrate scope design.
Reinforced Genetic Algorithm for Structure-based Drug Design
Nov 28, 2022


Structure-based drug design (SBDD) aims to discover drug candidates by finding molecules (ligands) that bind tightly to a disease-related protein (targets), which is the primary approach to computer-aided drug discovery. Recently, applying deep generative models for three-dimensional (3D) molecular design conditioned on protein pockets to solve SBDD has attracted much attention, but their formulation as probabilistic modeling often leads to unsatisfactory optimization performance. On the other hand, traditional combinatorial optimization methods such as genetic algorithms (GA) have demonstrated state-of-the-art performance in various molecular optimization tasks. However, they do not utilize protein target structure to inform design steps but rely on a random-walk-like exploration, which leads to unstable performance and no knowledge transfer between different tasks despite the similar binding physics. To achieve a more stable and efficient SBDD, we propose Reinforced Genetic Algorithm (RGA) that uses neural models to prioritize the profitable design steps and suppress random-walk behavior. The neural models take the 3D structure of the targets and ligands as inputs and are pre-trained using native complex structures to utilize the knowledge of the shared binding physics from different targets and then fine-tuned during optimization. We conduct thorough empirical studies on optimizing binding affinity to various disease targets and show that RGA outperforms the baselines in terms of docking scores and is more robust to random initializations. The ablation study also indicates that the training on different targets helps improve performance by leveraging the shared underlying physics of the binding processes. The code is available at https://github.com/futianfan/reinforced-genetic-algorithm.
Amortized Tree Generation for Bottom-up Synthesis Planning and Synthesizable Molecular Design
Oct 12, 2021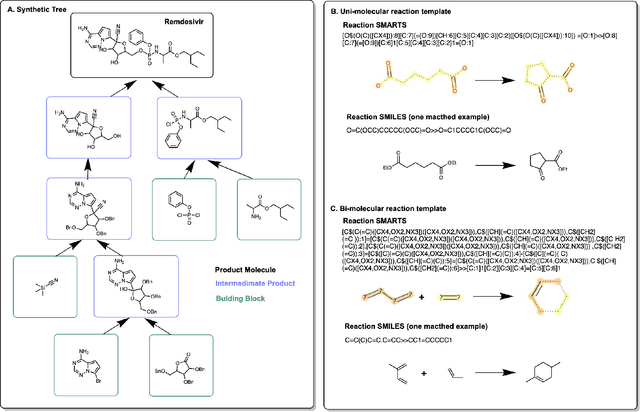
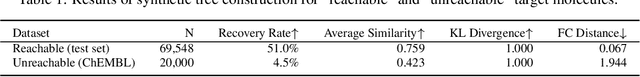
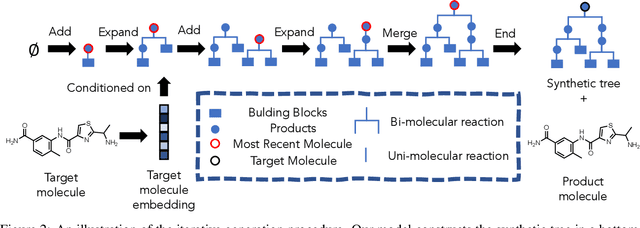
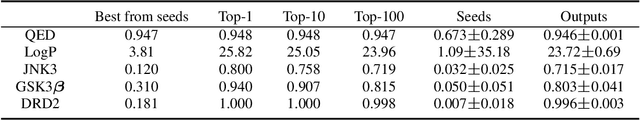
Molecular design and synthesis planning are two critical steps in the process of molecular discovery that we propose to formulate as a single shared task of conditional synthetic pathway generation. We report an amortized approach to generate synthetic pathways as a Markov decision process conditioned on a target molecular embedding. This approach allows us to conduct synthesis planning in a bottom-up manner and design synthesizable molecules by decoding from optimized conditional codes, demonstrating the potential to solve both problems of design and synthesis simultaneously. The approach leverages neural networks to probabilistically model the synthetic trees, one reaction step at a time, according to reactivity rules encoded in a discrete action space of reaction templates. We train these networks on hundreds of thousands of artificial pathways generated from a pool of purchasable compounds and a list of expert-curated templates. We validate our method with (a) the recovery of molecules using conditional generation, (b) the identification of synthesizable structural analogs, and (c) the optimization of molecular structures given oracle functions relevant to drug discovery.
Differentiable Scaffolding Tree for Molecular Optimization
Sep 22, 2021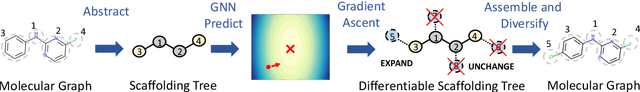

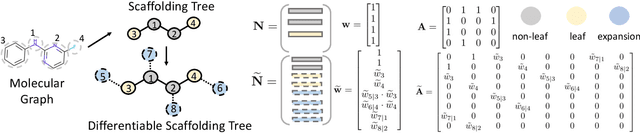
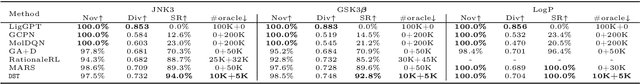
The structural design of functional molecules, also called molecular optimization, is an essential chemical science and engineering task with important applications, such as drug discovery. Deep generative models and combinatorial optimization methods achieve initial success but still struggle with directly modeling discrete chemical structures and often heavily rely on brute-force enumeration. The challenge comes from the discrete and non-differentiable nature of molecule structures. To address this, we propose differentiable scaffolding tree (DST) that utilizes a learned knowledge network to convert discrete chemical structures to locally differentiable ones. DST enables a gradient-based optimization on a chemical graph structure by back-propagating the derivatives from the target properties through a graph neural network (GNN). Our empirical studies show the gradient-based molecular optimizations are both effective and sample efficient. Furthermore, the learned graph parameters can also provide an explanation that helps domain experts understand the model output.