 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAUTODIFF: Autoregressive Diffusion Modeling for Structure-based Drug Design
Apr 03, 2024Structure-based drug design (SBDD), which aims to generate molecules that can bind tightly to the target protein, is an essential problem in drug discovery, and previous approaches have achieved initial success. However, most existing methods still suffer from invalid local structure or unrealistic conformation issues, which are mainly due to the poor leaning of bond angles or torsional angles. To alleviate these problems, we propose AUTODIFF, a diffusion-based fragment-wise autoregressive generation model. Specifically, we design a novel molecule assembly strategy named conformal motif that preserves the conformation of local structures of molecules first, then we encode the interaction of the protein-ligand complex with an SE(3)-equivariant convolutional network and generate molecules motif-by-motif with diffusion modeling. In addition, we also improve the evaluation framework of SBDD by constraining the molecular weights of the generated molecules in the same range, together with some new metrics, which make the evaluation more fair and practical. Extensive experiments on CrossDocked2020 demonstrate that our approach outperforms the existing models in generating realistic molecules with valid structures and conformations while maintaining high binding affinity.
Molecular Attributes Transfer from Non-Parallel Data
Nov 30, 2021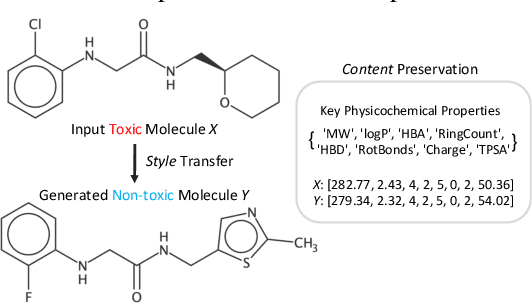
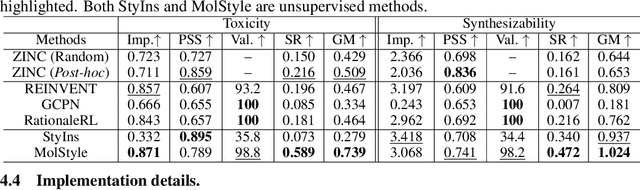
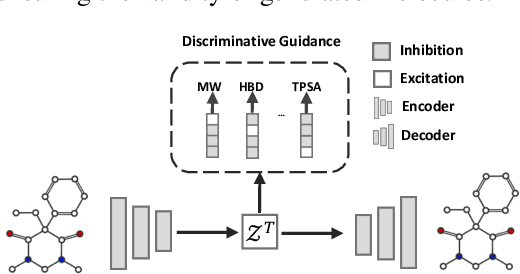
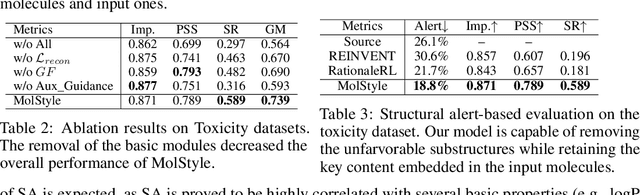
Optimizing chemical molecules for desired properties lies at the core of drug development. Despite initial successes made by deep generative models and reinforcement learning methods, these methods were mostly limited by the requirement of predefined attribute functions or parallel data with manually pre-compiled pairs of original and optimized molecules. In this paper, for the first time, we formulate molecular optimization as a style transfer problem and present a novel generative model that could automatically learn internal differences between two groups of non-parallel data through adversarial training strategies. Our model further enables both preservation of molecular contents and optimization of molecular properties through combining auxiliary guided-variational autoencoders and generative flow techniques. Experiments on two molecular optimization tasks, toxicity modification and synthesizability improvement, demonstrate that our model significantly outperforms several state-of-the-art methods.
BioNavi-NP: Biosynthesis Navigator for Natural Products
May 26, 2021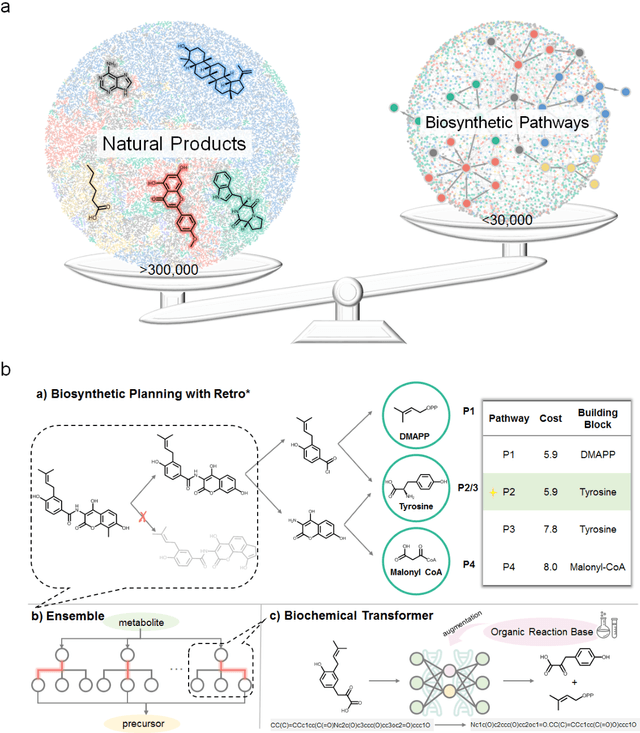
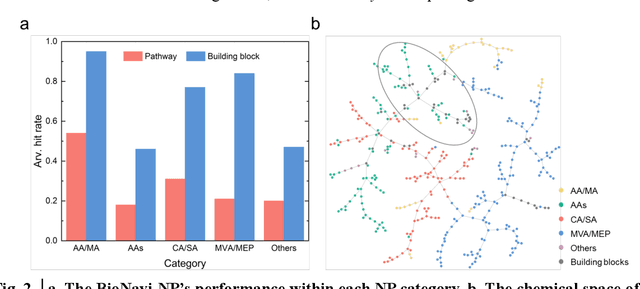
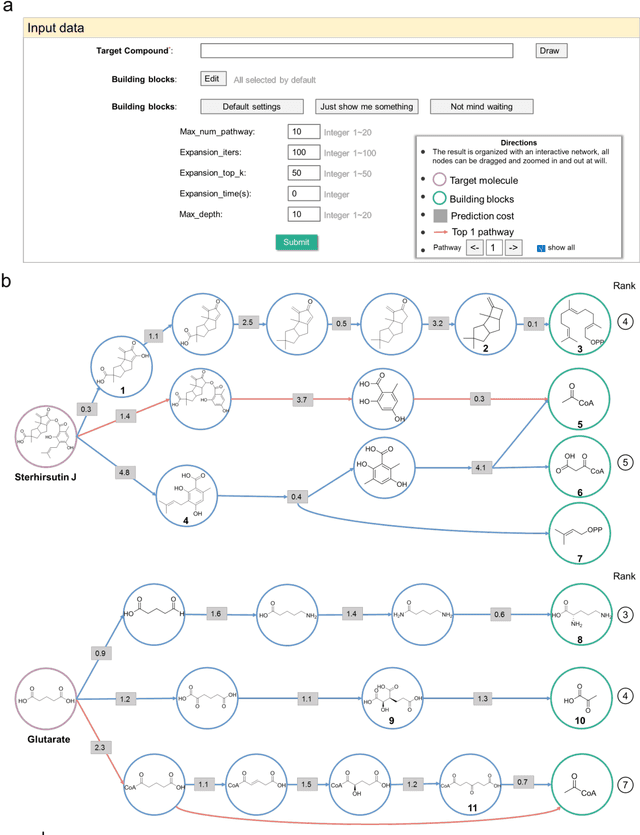
Nature, a synthetic master, creates more than 300,000 natural products (NPs) which are the major constituents of FDA-proved drugs owing to the vast chemical space of NPs. To date, there are fewer than 30,000 validated NPs compounds involved in about 33,000 known enzyme catalytic reactions, and even fewer biosynthetic pathways are known with complete cascade-connected enzyme catalysis. Therefore, it is valuable to make computer-aided bio-retrosynthesis predictions. Here, we develop BioNavi-NP, a navigable and user-friendly toolkit, which is capable of predicting the biosynthetic pathways for NPs and NP-like compounds through a novel (AND-OR Tree)-based planning algorithm, an enhanced molecular Transformer neural network, and a training set that combines general organic transformations and biosynthetic steps. Extensive evaluations reveal that BioNavi-NP generalizes well to identifying the reported biosynthetic pathways for 90% of test compounds and recovering the verified building blocks for 73%, significantly outperforming conventional rule-based approaches. Moreover, BioNavi-NP also shows an outstanding capacity of biologically plausible pathways enumeration. In this sense, BioNavi-NP is a leading-edge toolkit to redesign complex biosynthetic pathways of natural products with applications to total or semi-synthesis and pathway elucidation or reconstruction.
Retro*: Learning Retrosynthetic Planning with Neural Guided A* Search
Jun 29, 2020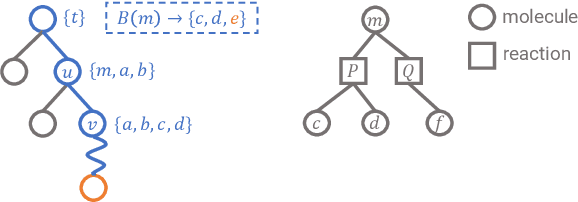
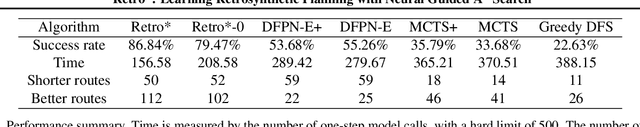
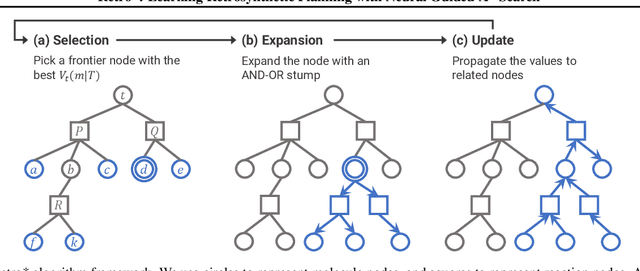
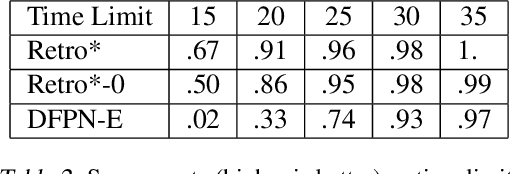
Retrosynthetic planning is a critical task in organic chemistry which identifies a series of reactions that can lead to the synthesis of a target product. The vast number of possible chemical transformations makes the size of the search space very big, and retrosynthetic planning is challenging even for experienced chemists. However, existing methods either require expensive return estimation by rollout with high variance, or optimize for search speed rather than the quality. In this paper, we propose Retro*, a neural-based A*-like algorithm that finds high-quality synthetic routes efficiently. It maintains the search as an AND-OR tree, and learns a neural search bias with off-policy data. Then guided by this neural network, it performs best-first search efficiently during new planning episodes. Experiments on benchmark USPTO datasets show that, our proposed method outperforms existing state-of-the-art with respect to both the success rate and solution quality, while being more efficient at the same time.
Retrosynthesis Prediction with Conditional Graph Logic Network
Jan 06, 2020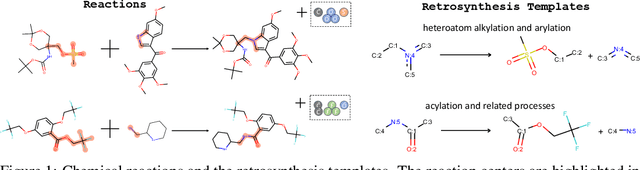
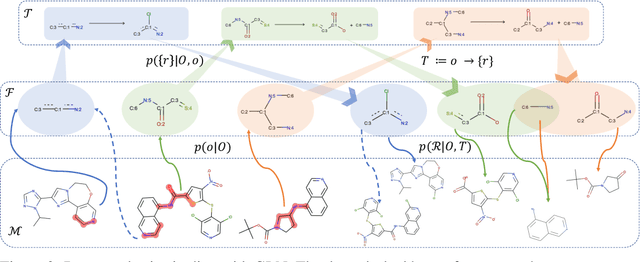
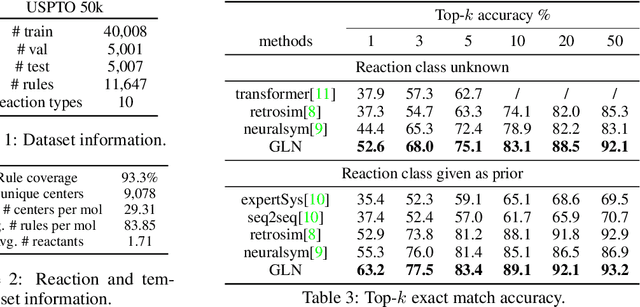
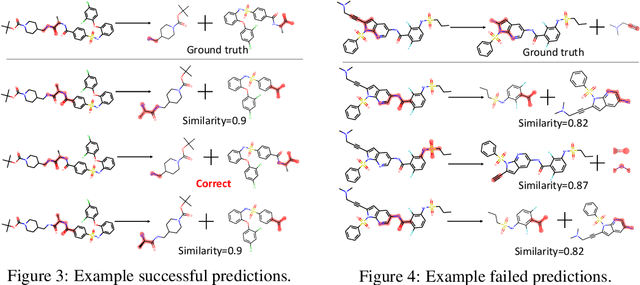
Retrosynthesis is one of the fundamental problems in organic chemistry. The task is to identify reactants that can be used to synthesize a specified product molecule. Recently, computer-aided retrosynthesis is finding renewed interest from both chemistry and computer science communities. Most existing approaches rely on template-based models that define subgraph matching rules, but whether or not a chemical reaction can proceed is not defined by hard decision rules. In this work, we propose a new approach to this task using the Conditional Graph Logic Network, a conditional graphical model built upon graph neural networks that learns when rules from reaction templates should be applied, implicitly considering whether the resulting reaction would be both chemically feasible and strategic. We also propose an efficient hierarchical sampling to alleviate the computation cost. While achieving a significant improvement of $8.1\%$ over current state-of-the-art methods on the benchmark dataset, our model also offers interpretations for the prediction.
Improving Sequential Determinantal Point Processes for Supervised Video Summarization
Oct 24, 2018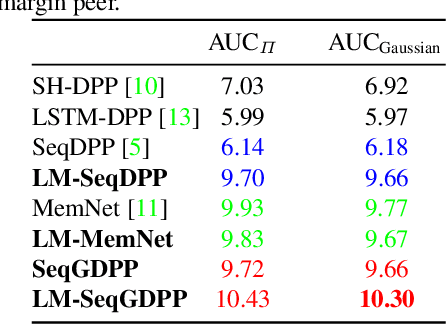
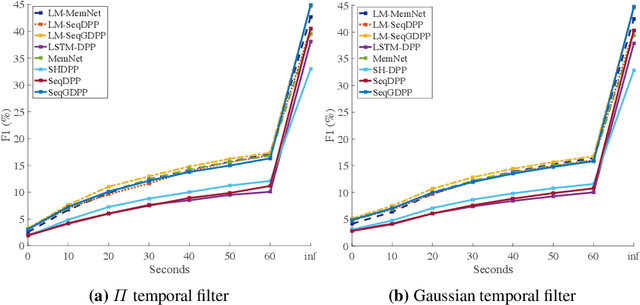
It is now much easier than ever before to produce videos. While the ubiquitous video data is a great source for information discovery and extraction, the computational challenges are unparalleled. Automatically summarizing the videos has become a substantial need for browsing, searching, and indexing visual content. This paper is in the vein of supervised video summarization using sequential determinantal point process (SeqDPP), which models diversity by a probabilistic distribution. We improve this model in two folds. In terms of learning, we propose a large-margin algorithm to address the exposure bias problem in SeqDPP. In terms of modeling, we design a new probabilistic distribution such that, when it is integrated into SeqDPP, the resulting model accepts user input about the expected length of the summary. Moreover, we also significantly extend a popular video summarization dataset by 1) more egocentric videos, 2) dense user annotations, and 3) a refined evaluation scheme. We conduct extensive experiments on this dataset (about 60 hours of videos in total) and compare our approach to several competitive baselines.
Representation Learning on Graphs with Jumping Knowledge Networks
Jun 25, 2018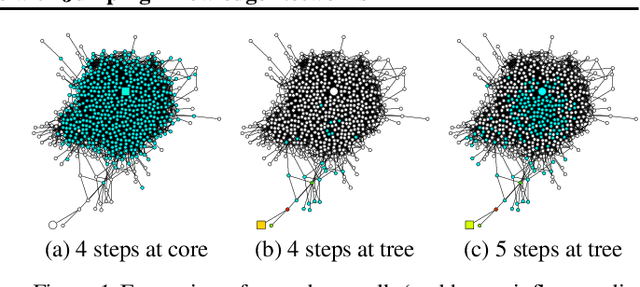
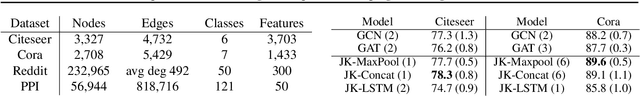
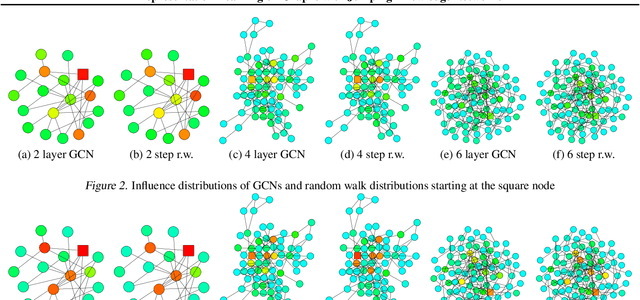
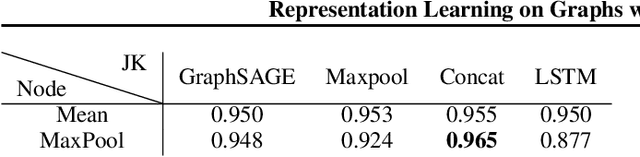
Recent deep learning approaches for representation learning on graphs follow a neighborhood aggregation procedure. We analyze some important properties of these models, and propose a strategy to overcome those. In particular, the range of "neighboring" nodes that a node's representation draws from strongly depends on the graph structure, analogous to the spread of a random walk. To adapt to local neighborhood properties and tasks, we explore an architecture -- jumping knowledge (JK) networks -- that flexibly leverages, for each node, different neighborhood ranges to enable better structure-aware representation. In a number of experiments on social, bioinformatics and citation networks, we demonstrate that our model achieves state-of-the-art performance. Furthermore, combining the JK framework with models like Graph Convolutional Networks, GraphSAGE and Graph Attention Networks consistently improves those models' performance.
Robust GANs against Dishonest Adversaries
Feb 27, 2018
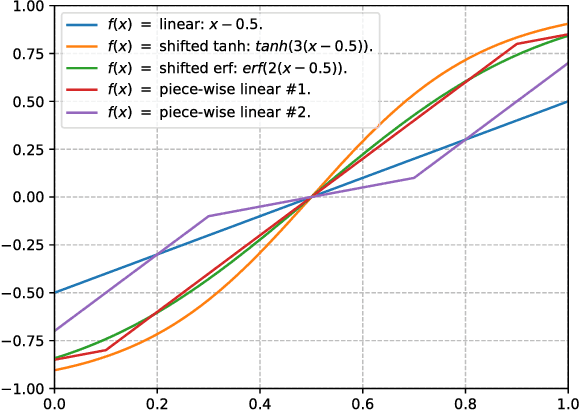
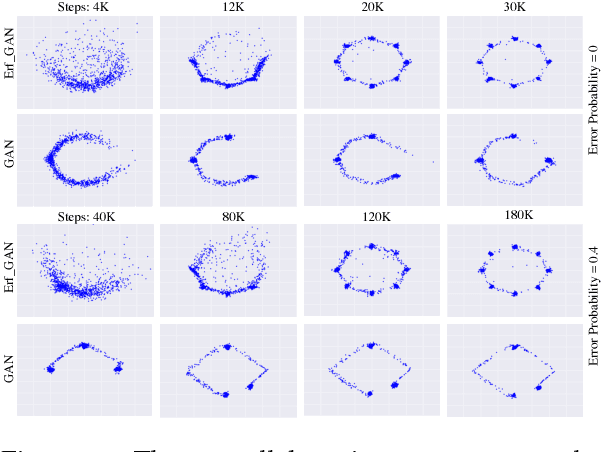
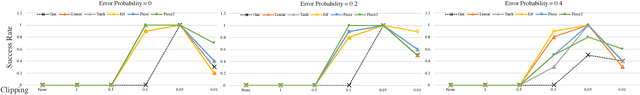
Robustness of deep learning models is a property that has recently gained increasing attention. We formally define a notion of robustness for generative adversarial models, and show that, perhaps surprisingly, the GAN in its original form is not robust. Indeed, the discriminator in GANs may be viewed as merely offering "teaching feedback". Our notion of robustness relies on a dishonest discriminator, or noisy, adversarial interference with its feedback. We explore, theoretically and empirically, the effect of model and training properties on this robustness. In particular, we show theoretical conditions for robustness that are supported by empirical evidence. We also test the effect of regularization. Our results suggest variations of GANs that are indeed more robust to noisy attacks, and have overall more stable training behavior.
Batched High-dimensional Bayesian Optimization via Structural Kernel Learning
Jan 06, 2018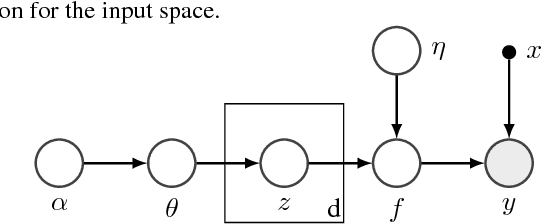
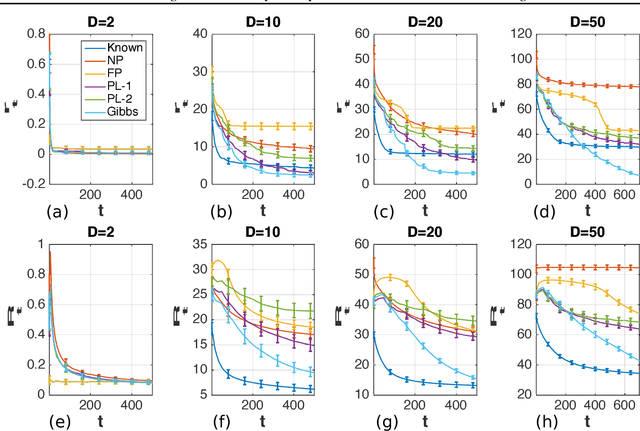
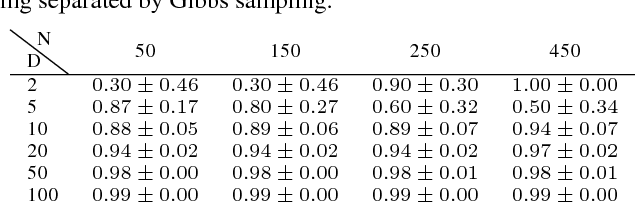
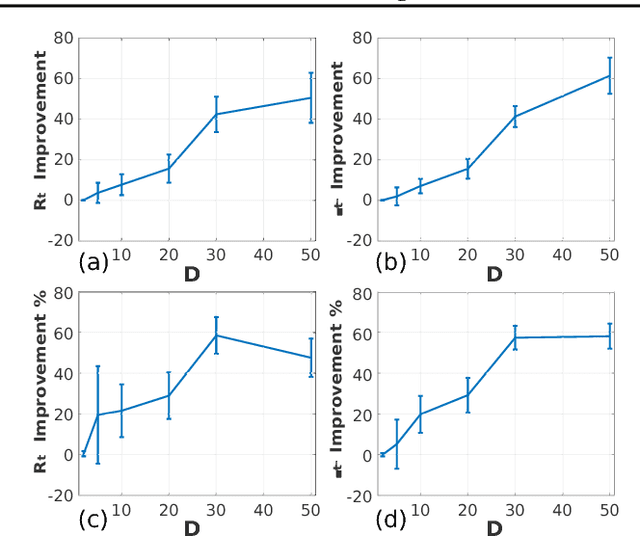
Optimization of high-dimensional black-box functions is an extremely challenging problem. While Bayesian optimization has emerged as a popular approach for optimizing black-box functions, its applicability has been limited to low-dimensional problems due to its computational and statistical challenges arising from high-dimensional settings. In this paper, we propose to tackle these challenges by (1) assuming a latent additive structure in the function and inferring it properly for more efficient and effective BO, and (2) performing multiple evaluations in parallel to reduce the number of iterations required by the method. Our novel approach learns the latent structure with Gibbs sampling and constructs batched queries using determinantal point processes. Experimental validations on both synthetic and real-world functions demonstrate that the proposed method outperforms the existing state-of-the-art approaches.
Polynomial Time Algorithms for Dual Volume Sampling
Nov 16, 2017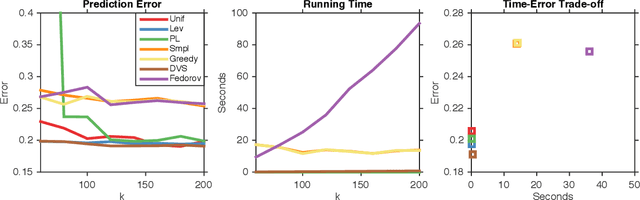
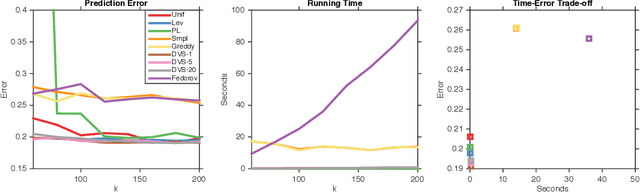
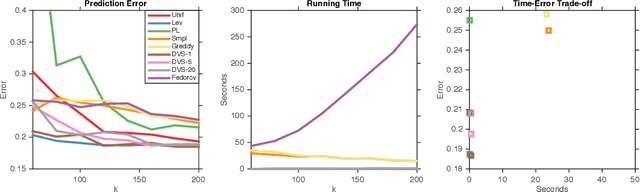
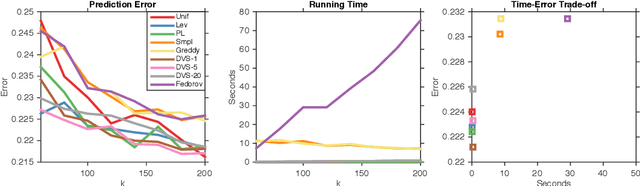
We study dual volume sampling, a method for selecting k columns from an n x m short and wide matrix (n <= k <= m) such that the probability of selection is proportional to the volume spanned by the rows of the induced submatrix. This method was proposed by Avron and Boutsidis (2013), who showed it to be a promising method for column subset selection and its multiple applications. However, its wider adoption has been hampered by the lack of polynomial time sampling algorithms. We remove this hindrance by developing an exact (randomized) polynomial time sampling algorithm as well as its derandomization. Thereafter, we study dual volume sampling via the theory of real stable polynomials and prove that its distribution satisfies the "Strong Rayleigh" property. This result has numerous consequences, including a provably fast-mixing Markov chain sampler that makes dual volume sampling much more attractive to practitioners. This sampler is closely related to classical algorithms for popular experimental design methods that are to date lacking theoretical analysis but are known to empirically work well.