 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePixCell: A generative foundation model for digital histopathology images
Jun 05, 2025The digitization of histology slides has revolutionized pathology, providing massive datasets for cancer diagnosis and research. Contrastive self-supervised and vision-language models have been shown to effectively mine large pathology datasets to learn discriminative representations. On the other hand, generative models, capable of synthesizing realistic and diverse images, present a compelling solution to address unique problems in pathology that involve synthesizing images; overcoming annotated data scarcity, enabling privacy-preserving data sharing, and performing inherently generative tasks, such as virtual staining. We introduce PixCell, the first diffusion-based generative foundation model for histopathology. We train PixCell on PanCan-30M, a vast, diverse dataset derived from 69,184 H\&E-stained whole slide images covering various cancer types. We employ a progressive training strategy and a self-supervision-based conditioning that allows us to scale up training without any annotated data. PixCell generates diverse and high-quality images across multiple cancer types, which we find can be used in place of real data to train a self-supervised discriminative model. Synthetic images shared between institutions are subject to fewer regulatory barriers than would be the case with real clinical images. Furthermore, we showcase the ability to precisely control image generation using a small set of annotated images, which can be used for both data augmentation and educational purposes. Testing on a cell segmentation task, a mask-guided PixCell enables targeted data augmentation, improving downstream performance. Finally, we demonstrate PixCell's ability to use H\&E structural staining to infer results from molecular marker studies; we use this capability to infer IHC staining from H\&E images. Our trained models are publicly released to accelerate research in computational pathology.
RankByGene: Gene-Guided Histopathology Representation Learning Through Cross-Modal Ranking Consistency
Nov 22, 2024Spatial transcriptomics (ST) provides essential spatial context by mapping gene expression within tissue, enabling detailed study of cellular heterogeneity and tissue organization. However, aligning ST data with histology images poses challenges due to inherent spatial distortions and modality-specific variations. Existing methods largely rely on direct alignment, which often fails to capture complex cross-modal relationships. To address these limitations, we propose a novel framework that aligns gene and image features using a ranking-based alignment loss, preserving relative similarity across modalities and enabling robust multi-scale alignment. To further enhance the alignment's stability, we employ self-supervised knowledge distillation with a teacher-student network architecture, effectively mitigating disruptions from high dimensionality, sparsity, and noise in gene expression data. Extensive experiments on gene expression prediction and survival analysis demonstrate our framework's effectiveness, showing improved alignment and predictive performance over existing methods and establishing a robust tool for gene-guided image representation learning in digital pathology.
Open and reusable deep learning for pathology with WSInfer and QuPath
Sep 08, 2023The field of digital pathology has seen a proliferation of deep learning models in recent years. Despite substantial progress, it remains rare for other researchers and pathologists to be able to access models published in the literature and apply them to their own images. This is due to difficulties in both sharing and running models. To address these concerns, we introduce WSInfer: a new, open-source software ecosystem designed to make deep learning for pathology more streamlined and accessible. WSInfer comprises three main elements: 1) a Python package and command line tool to efficiently apply patch-based deep learning inference to whole slide images; 2) a QuPath extension that provides an alternative inference engine through user-friendly and interactive software, and 3) a model zoo, which enables pathology models and metadata to be easily shared in a standardized form. Together, these contributions aim to encourage wider reuse, exploration, and interrogation of deep learning models for research purposes, by putting them into the hands of pathologists and eliminating a need for coding experience when accessed through QuPath. The WSInfer source code is hosted on GitHub and documentation is available at https://wsinfer.readthedocs.io.
PathLDM: Text conditioned Latent Diffusion Model for Histopathology
Sep 01, 2023



To achieve high-quality results, diffusion models must be trained on large datasets. This can be notably prohibitive for models in specialized domains, such as computational pathology. Conditioning on labeled data is known to help in data-efficient model training. Therefore, histopathology reports, which are rich in valuable clinical information, are an ideal choice as guidance for a histopathology generative model. In this paper, we introduce PathLDM, the first text-conditioned Latent Diffusion Model tailored for generating high-quality histopathology images. Leveraging the rich contextual information provided by pathology text reports, our approach fuses image and textual data to enhance the generation process. By utilizing GPT's capabilities to distill and summarize complex text reports, we establish an effective conditioning mechanism. Through strategic conditioning and necessary architectural enhancements, we achieved a SoTA FID score of 7.64 for text-to-image generation on the TCGA-BRCA dataset, significantly outperforming the closest text-conditioned competitor with FID 30.1.
Topology-Guided Multi-Class Cell Context Generation for Digital Pathology
Apr 05, 2023



In digital pathology, the spatial context of cells is important for cell classification, cancer diagnosis and prognosis. To model such complex cell context, however, is challenging. Cells form different mixtures, lineages, clusters and holes. To model such structural patterns in a learnable fashion, we introduce several mathematical tools from spatial statistics and topological data analysis. We incorporate such structural descriptors into a deep generative model as both conditional inputs and a differentiable loss. This way, we are able to generate high quality multi-class cell layouts for the first time. We show that the topology-rich cell layouts can be used for data augmentation and improve the performance of downstream tasks such as cell classification.
Understanding metric-related pitfalls in image analysis validation
Feb 09, 2023Validation metrics are key for the reliable tracking of scientific progress and for bridging the current chasm between artificial intelligence (AI) research and its translation into practice. However, increasing evidence shows that particularly in image analysis, metrics are often chosen inadequately in relation to the underlying research problem. This could be attributed to a lack of accessibility of metric-related knowledge: While taking into account the individual strengths, weaknesses, and limitations of validation metrics is a critical prerequisite to making educated choices, the relevant knowledge is currently scattered and poorly accessible to individual researchers. Based on a multi-stage Delphi process conducted by a multidisciplinary expert consortium as well as extensive community feedback, the present work provides the first reliable and comprehensive common point of access to information on pitfalls related to validation metrics in image analysis. Focusing on biomedical image analysis but with the potential of transfer to other fields, the addressed pitfalls generalize across application domains and are categorized according to a newly created, domain-agnostic taxonomy. To facilitate comprehension, illustrations and specific examples accompany each pitfall. As a structured body of information accessible to researchers of all levels of expertise, this work enhances global comprehension of a key topic in image analysis validation.
Learning Topological Interactions for Multi-Class Medical Image Segmentation
Jul 20, 2022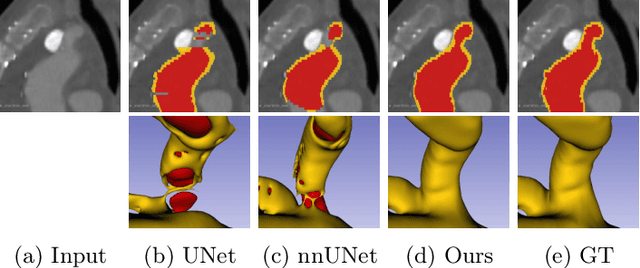
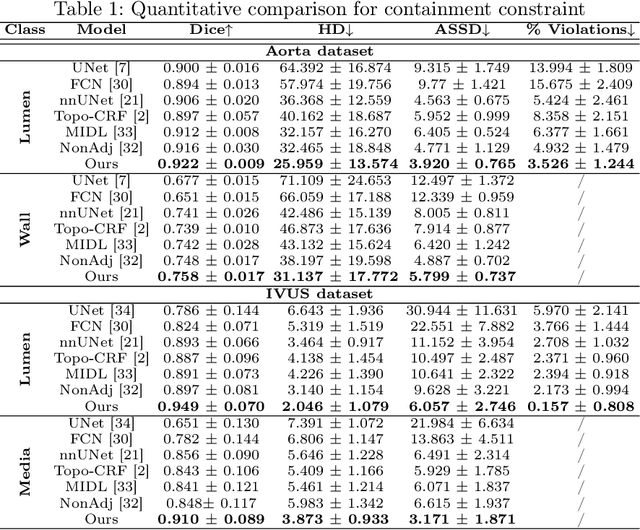
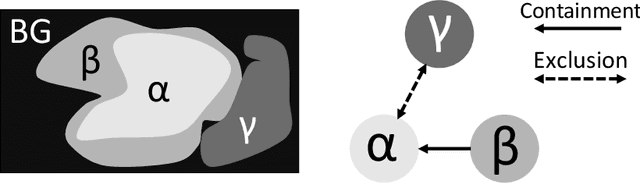
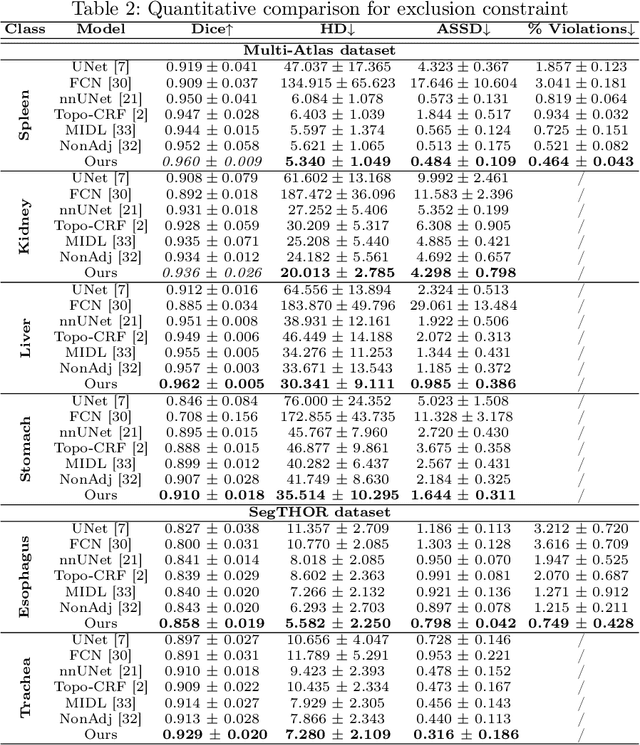
Deep learning methods have achieved impressive performance for multi-class medical image segmentation. However, they are limited in their ability to encode topological interactions among different classes (e.g., containment and exclusion). These constraints naturally arise in biomedical images and can be crucial in improving segmentation quality. In this paper, we introduce a novel topological interaction module to encode the topological interactions into a deep neural network. The implementation is completely convolution-based and thus can be very efficient. This empowers us to incorporate the constraints into end-to-end training and enrich the feature representation of neural networks. The efficacy of the proposed method is validated on different types of interactions. We also demonstrate the generalizability of the method on both proprietary and public challenge datasets, in both 2D and 3D settings, as well as across different modalities such as CT and Ultrasound. Code is available at: https://github.com/TopoXLab/TopoInteraction
AI and Pathology: Steering Treatment and Predicting Outcomes
Jun 15, 2022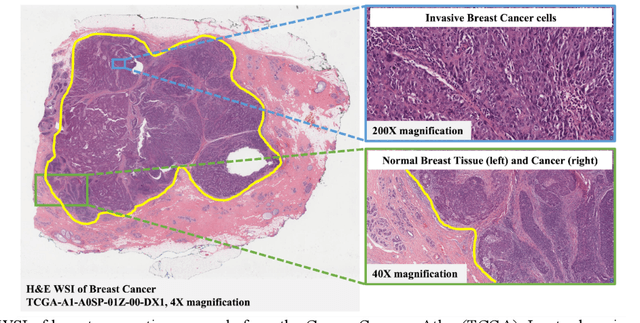
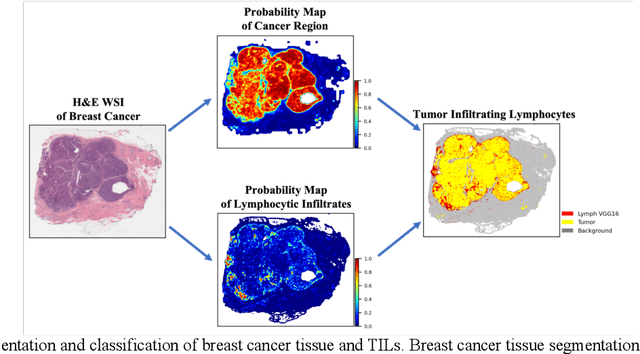
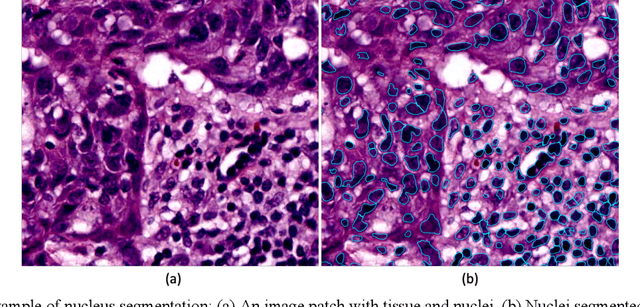
The combination of data analysis methods, increasing computing capacity, and improved sensors enable quantitative granular, multi-scale, cell-based analyses. We describe the rich set of application challenges related to tissue interpretation and survey AI methods currently used to address these challenges. We focus on a particular class of targeted human tissue analysis - histopathology - aimed at quantitative characterization of disease state, patient outcome prediction and treatment steering.
Metrics reloaded: Pitfalls and recommendations for image analysis validation
Jun 03, 2022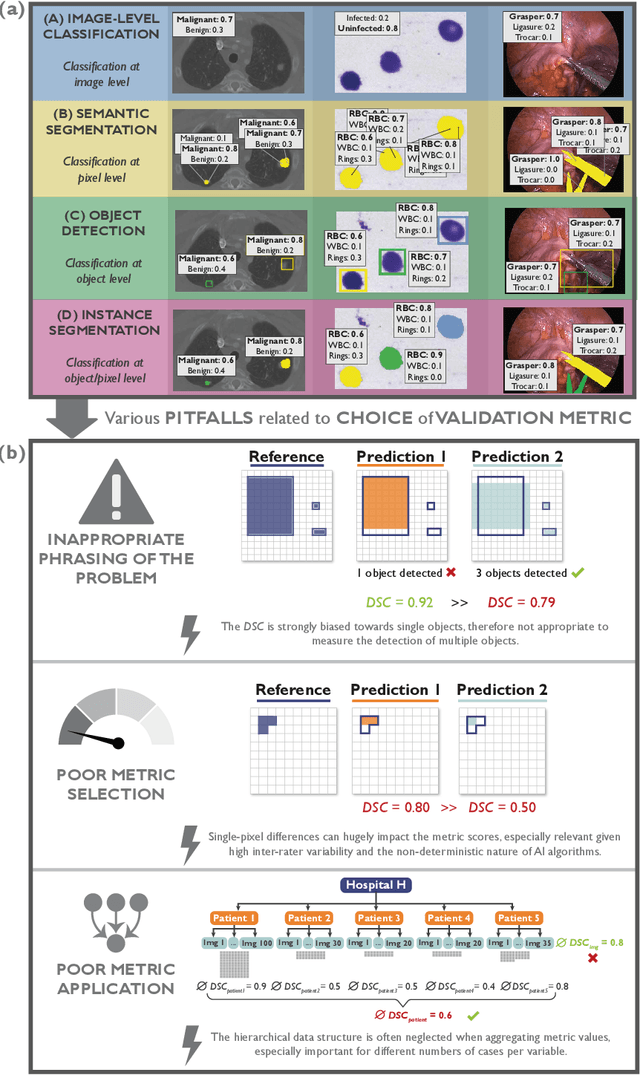
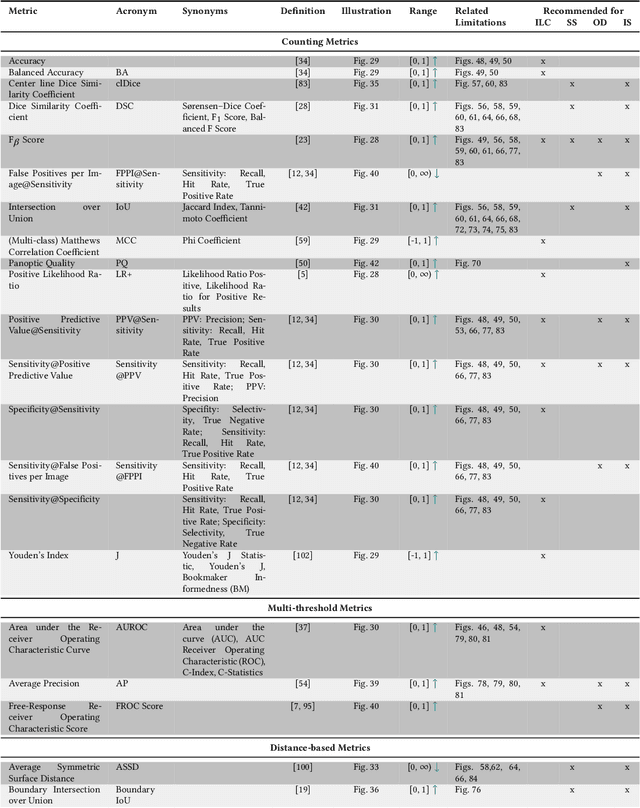
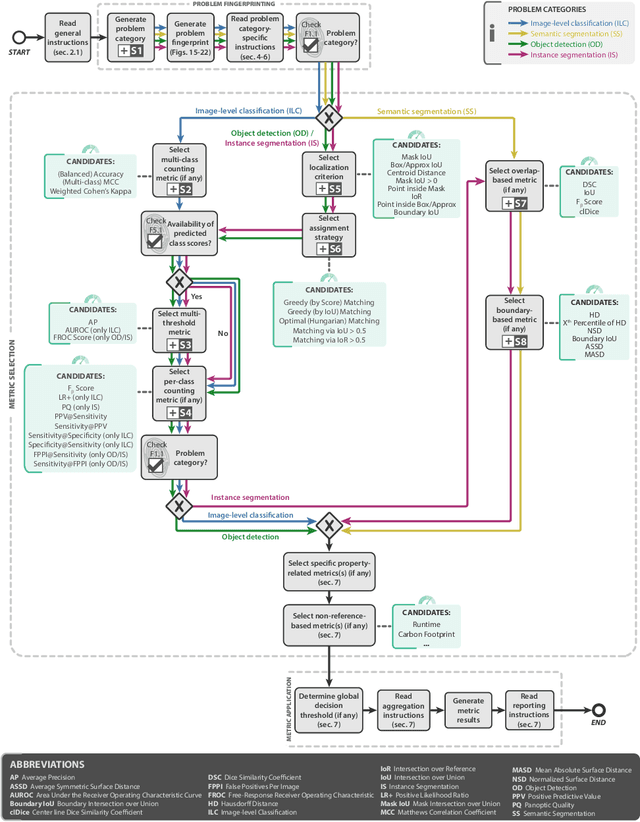
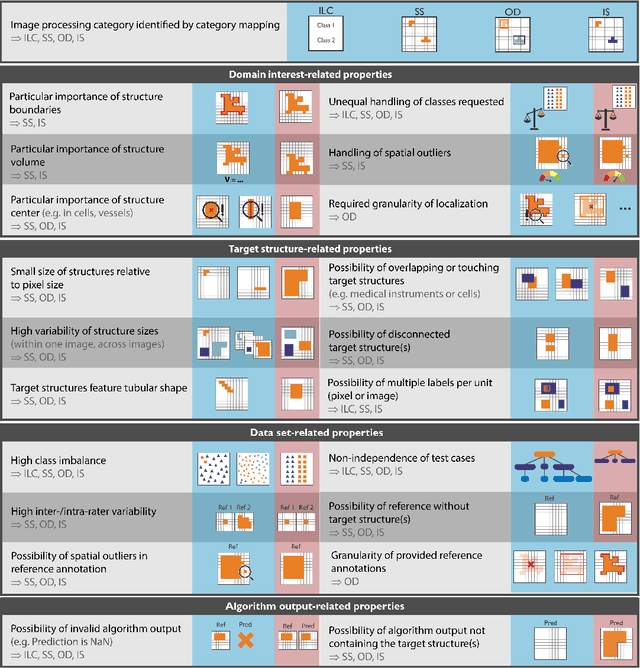
The field of automatic biomedical image analysis crucially depends on robust and meaningful performance metrics for algorithm validation. Current metric usage, however, is often ill-informed and does not reflect the underlying domain interest. Here, we present a comprehensive framework that guides researchers towards choosing performance metrics in a problem-aware manner. Specifically, we focus on biomedical image analysis problems that can be interpreted as a classification task at image, object or pixel level. The framework first compiles domain interest-, target structure-, data set- and algorithm output-related properties of a given problem into a problem fingerprint, while also mapping it to the appropriate problem category, namely image-level classification, semantic segmentation, instance segmentation, or object detection. It then guides users through the process of selecting and applying a set of appropriate validation metrics while making them aware of potential pitfalls related to individual choices. In this paper, we describe the current status of the Metrics Reloaded recommendation framework, with the goal of obtaining constructive feedback from the image analysis community. The current version has been developed within an international consortium of more than 60 image analysis experts and will be made openly available as a user-friendly toolkit after community-driven optimization.
A Novel Framework for Characterization of Tumor-Immune Spatial Relationships in Tumor Microenvironment
May 01, 2022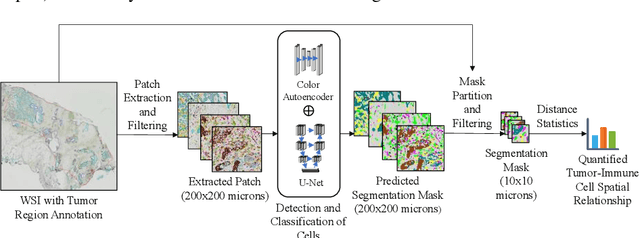

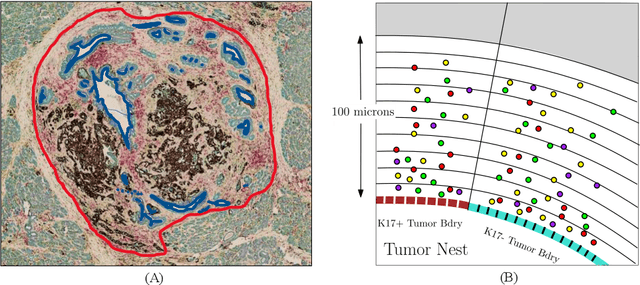
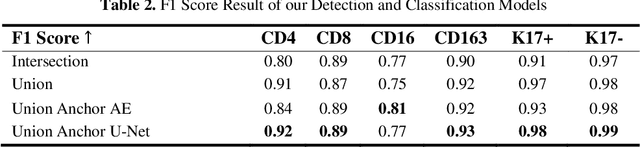
Understanding the impact of tumor biology on the composition of nearby cells often requires characterizing the impact of biologically distinct tumor regions. Biomarkers have been developed to label biologically distinct tumor regions, but challenges arise because of differences in the spatial extent and distribution of differentially labeled regions. In this work, we present a framework for systematically investigating the impact of distinct tumor regions on cells near the tumor borders, accounting their cross spatial distributions. We apply the framework to multiplex immunohistochemistry (mIHC) studies of pancreatic cancer and show its efficacy in demonstrating how biologically different tumor regions impact the immune response in the tumor microenvironment. Furthermore, we show that the proposed framework can be extended to largescale whole slide image analysis.