 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReusable specimen-level inference in computational pathology
Jan 10, 2025

Foundation models for computational pathology have shown great promise for specimen-level tasks and are increasingly accessible to researchers. However, specimen-level models built on these foundation models remain largely unavailable, hindering their broader utility and impact. To address this gap, we developed SpinPath, a toolkit designed to democratize specimen-level deep learning by providing a zoo of pretrained specimen-level models, a Python-based inference engine, and a JavaScript-based inference platform. We demonstrate the utility of SpinPath in metastasis detection tasks across nine foundation models. SpinPath may foster reproducibility, simplify experimentation, and accelerate the adoption of specimen-level deep learning in computational pathology research.
Decoding the visual attention of pathologists to reveal their level of expertise
Mar 25, 2024



We present a method for classifying the expertise of a pathologist based on how they allocated their attention during a cancer reading. We engage this decoding task by developing a novel method for predicting the attention of pathologists as they read whole-slide Images (WSIs) of prostate and make cancer grade classifications. Our ground truth measure of a pathologists' attention is the x, y and z (magnification) movement of their viewport as they navigated through WSIs during readings, and to date we have the attention behavior of 43 pathologists reading 123 WSIs. These data revealed that specialists have higher agreement in both their attention and cancer grades compared to general pathologists and residents, suggesting that sufficient information may exist in their attention behavior to classify their expertise level. To attempt this, we trained a transformer-based model to predict the visual attention heatmaps of resident, general, and specialist (GU) pathologists during Gleason grading. Based solely on a pathologist's attention during a reading, our model was able to predict their level of expertise with 75.3%, 56.1%, and 77.2% accuracy, respectively, better than chance and baseline models. Our model therefore enables a pathologist's expertise level to be easily and objectively evaluated, important for pathology training and competency assessment. Tools developed from our model could also be used to help pathology trainees learn how to read WSIs like an expert.
Open and reusable deep learning for pathology with WSInfer and QuPath
Sep 08, 2023The field of digital pathology has seen a proliferation of deep learning models in recent years. Despite substantial progress, it remains rare for other researchers and pathologists to be able to access models published in the literature and apply them to their own images. This is due to difficulties in both sharing and running models. To address these concerns, we introduce WSInfer: a new, open-source software ecosystem designed to make deep learning for pathology more streamlined and accessible. WSInfer comprises three main elements: 1) a Python package and command line tool to efficiently apply patch-based deep learning inference to whole slide images; 2) a QuPath extension that provides an alternative inference engine through user-friendly and interactive software, and 3) a model zoo, which enables pathology models and metadata to be easily shared in a standardized form. Together, these contributions aim to encourage wider reuse, exploration, and interrogation of deep learning models for research purposes, by putting them into the hands of pathologists and eliminating a need for coding experience when accessed through QuPath. The WSInfer source code is hosted on GitHub and documentation is available at https://wsinfer.readthedocs.io.
Evaluating histopathology transfer learning with ChampKit
Jun 14, 2022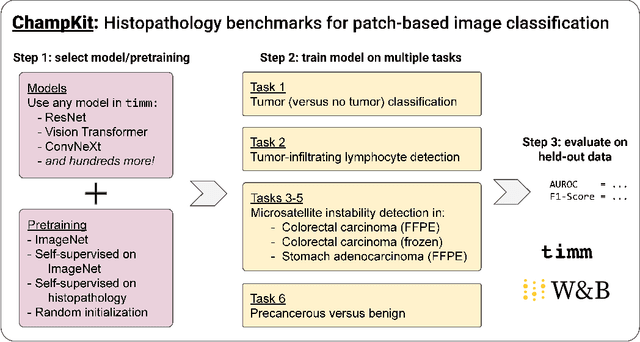
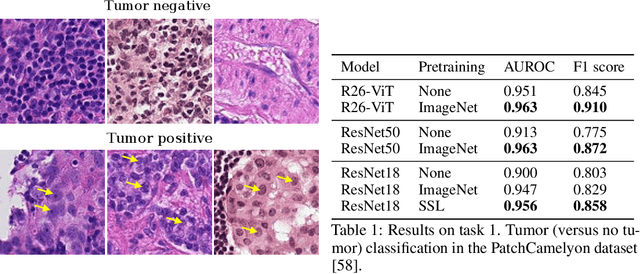
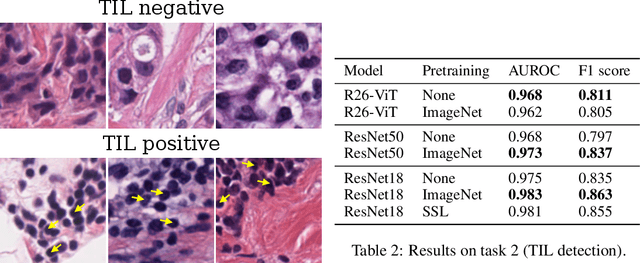
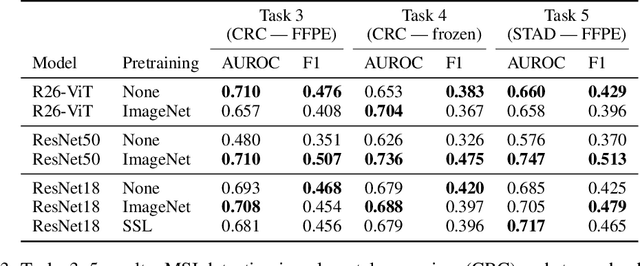
Histopathology remains the gold standard for diagnosis of various cancers. Recent advances in computer vision, specifically deep learning, have facilitated the analysis of histopathology images for various tasks, including immune cell detection and microsatellite instability classification. The state-of-the-art for each task often employs base architectures that have been pretrained for image classification on ImageNet. The standard approach to develop classifiers in histopathology tends to focus narrowly on optimizing models for a single task, not considering the aspects of modeling innovations that improve generalization across tasks. Here we present ChampKit (Comprehensive Histopathology Assessment of Model Predictions toolKit): an extensible, fully reproducible benchmarking toolkit that consists of a broad collection of patch-level image classification tasks across different cancers. ChampKit enables a way to systematically document the performance impact of proposed improvements in models and methodology. ChampKit source code and data are freely accessible at https://github.com/kaczmarj/champkit .
Federated Learning for the Classification of Tumor Infiltrating Lymphocytes
Apr 01, 2022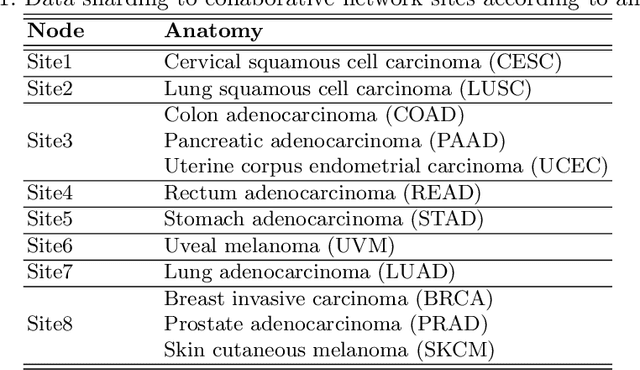
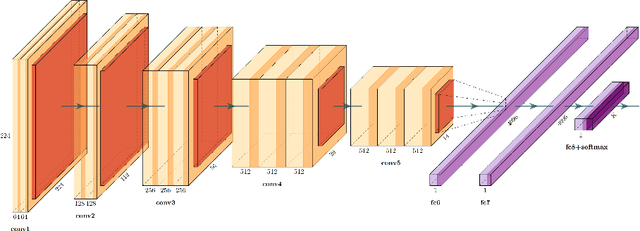
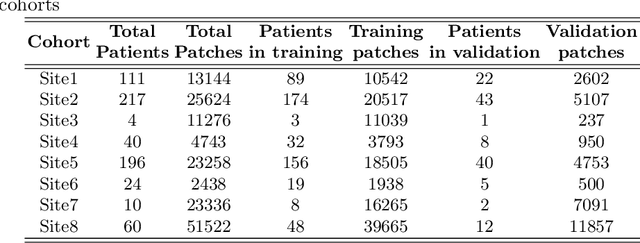

We evaluate the performance of federated learning (FL) in developing deep learning models for analysis of digitized tissue sections. A classification application was considered as the example use case, on quantifiying the distribution of tumor infiltrating lymphocytes within whole slide images (WSIs). A deep learning classification model was trained using 50*50 square micron patches extracted from the WSIs. We simulated a FL environment in which a dataset, generated from WSIs of cancer from numerous anatomical sites available by The Cancer Genome Atlas repository, is partitioned in 8 different nodes. Our results show that the model trained with the federated training approach achieves similar performance, both quantitatively and qualitatively, to that of a model trained with all the training data pooled at a centralized location. Our study shows that FL has tremendous potential for enabling development of more robust and accurate models for histopathology image analysis without having to collect large and diverse training data at a single location.
Visual attention analysis of pathologists examining whole slide images of Prostate cancer
Feb 17, 2022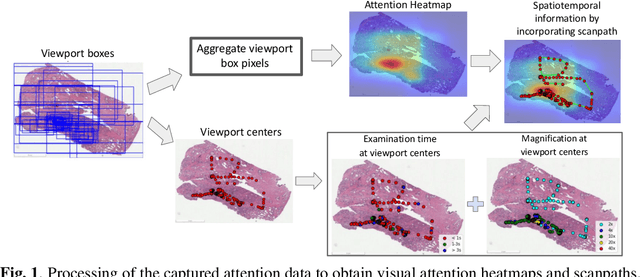
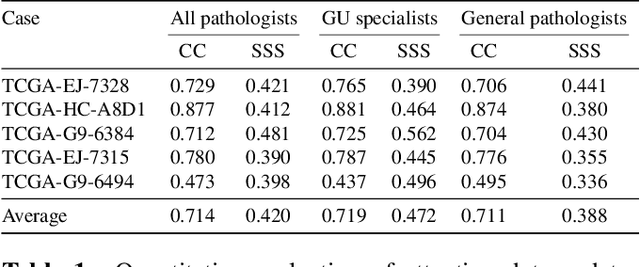
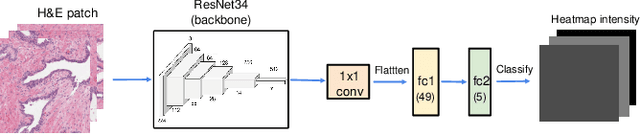
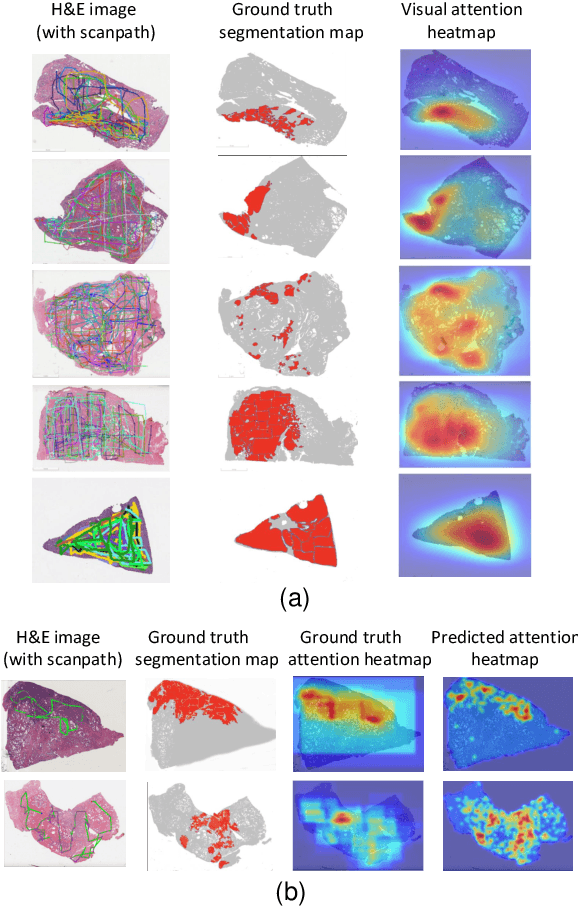
We study the attention of pathologists as they examine whole-slide images (WSIs) of prostate cancer tissue using a digital microscope. To the best of our knowledge, our study is the first to report in detail how pathologists navigate WSIs of prostate cancer as they accumulate information for their diagnoses. We collected slide navigation data (i.e., viewport location, magnification level, and time) from 13 pathologists in 2 groups (5 genitourinary (GU) specialists and 8 general pathologists) and generated visual attention heatmaps and scanpaths. Each pathologist examined five WSIs from the TCGA PRAD dataset, which were selected by a GU pathology specialist. We examined and analyzed the distributions of visual attention for each group of pathologists after each WSI was examined. To quantify the relationship between a pathologist's attention and evidence for cancer in the WSI, we obtained tumor annotations from a genitourinary specialist. We used these annotations to compute the overlap between the distribution of visual attention and annotated tumor region to identify strong correlations. Motivated by this analysis, we trained a deep learning model to predict visual attention on unseen WSIs. We find that the attention heatmaps predicted by our model correlate quite well with the ground truth attention heatmap and tumor annotations on a test set of 17 WSIs by using various spatial and temporal evaluation metrics.
Dataset of Segmented Nuclei in Hematoxylin and Eosin Stained Histopathology Images of 10 Cancer Types
Feb 18, 2020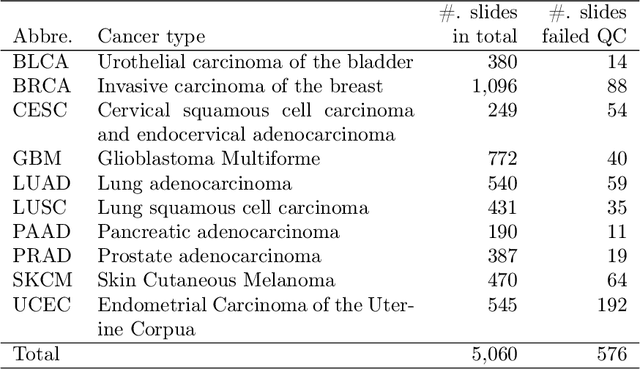
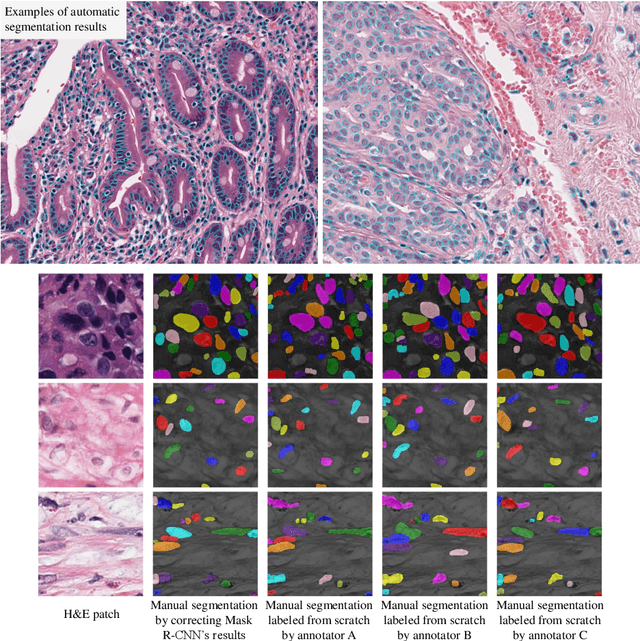
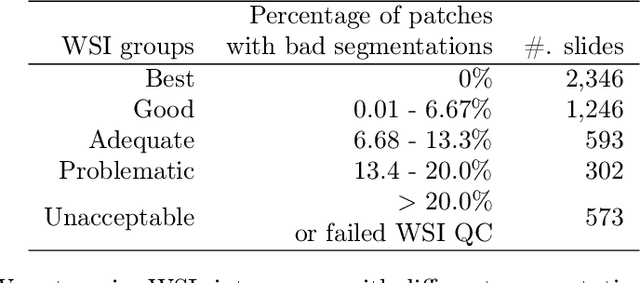
The distribution and appearance of nuclei are essential markers for the diagnosis and study of cancer. Despite the importance of nuclear morphology, there is a lack of large scale, accurate, publicly accessible nucleus segmentation data. To address this, we developed an analysis pipeline that segments nuclei in whole slide tissue images from multiple cancer types with a quality control process. We have generated nucleus segmentation results in 5,060 Whole Slide Tissue images from 10 cancer types in The Cancer Genome Atlas. One key component of our work is that we carried out a multi-level quality control process (WSI-level and image patch-level), to evaluate the quality of our segmentation results. The image patch-level quality control used manual segmentation ground truth data from 1,356 sampled image patches. The datasets we publish in this work consist of roughly 5 billion quality controlled nuclei from more than 5,060 TCGA WSIs from 10 different TCGA cancer types and 1,356 manually segmented TCGA image patches from the same 10 cancer types plus additional 4 cancer types.
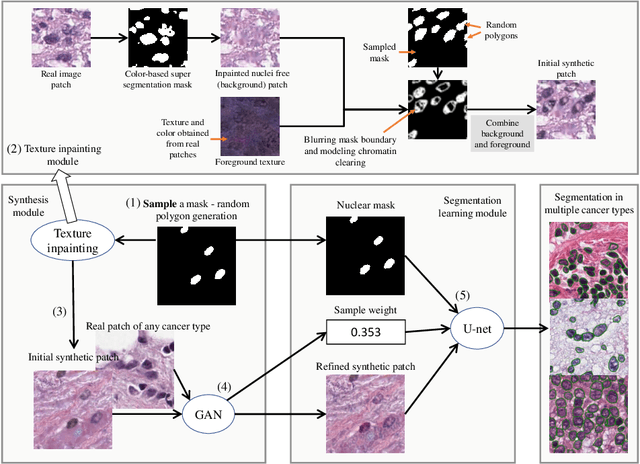
Unsupervised Histopathology Image Synthesis
Dec 13, 2017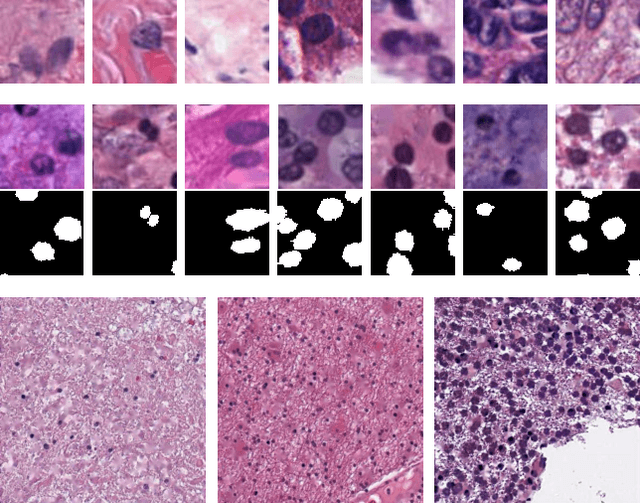
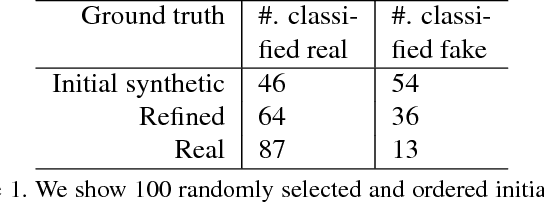
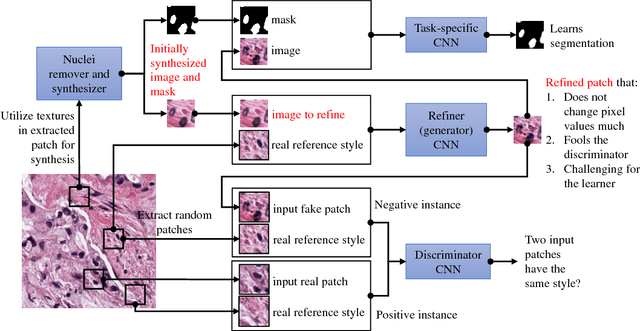
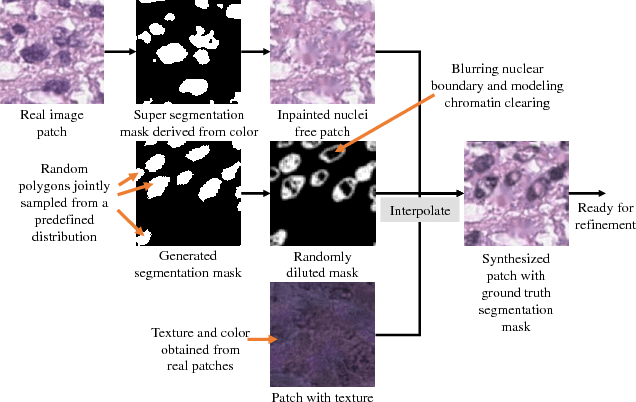
Hematoxylin and Eosin stained histopathology image analysis is essential for the diagnosis and study of complicated diseases such as cancer. Existing state-of-the-art approaches demand extensive amount of supervised training data from trained pathologists. In this work we synthesize in an unsupervised manner, large histopathology image datasets, suitable for supervised training tasks. We propose a unified pipeline that: a) generates a set of initial synthetic histopathology images with paired information about the nuclei such as segmentation masks; b) refines the initial synthetic images through a Generative Adversarial Network (GAN) to reference styles; c) trains a task-specific CNN and boosts the performance of the task-specific CNN with on-the-fly generated adversarial examples. Our main contribution is that the synthetic images are not only realistic, but also representative (in reference styles) and relatively challenging for training task-specific CNNs. We test our method for nucleus segmentation using images from four cancer types. When no supervised data exists for a cancer type, our method without supervision cost significantly outperforms supervised methods which perform across-cancer generalization. Even when supervised data exists for all cancer types, our approach without supervision cost performs better than supervised methods.
Sparse Autoencoder for Unsupervised Nucleus Detection and Representation in Histopathology Images
Apr 10, 2017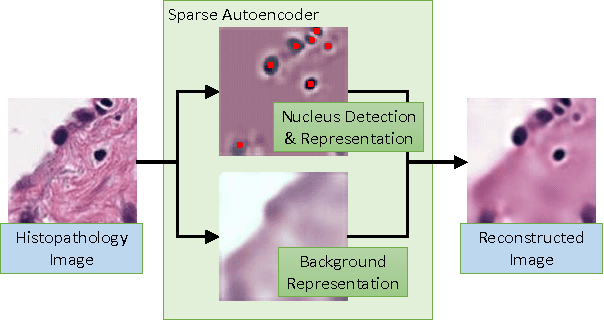
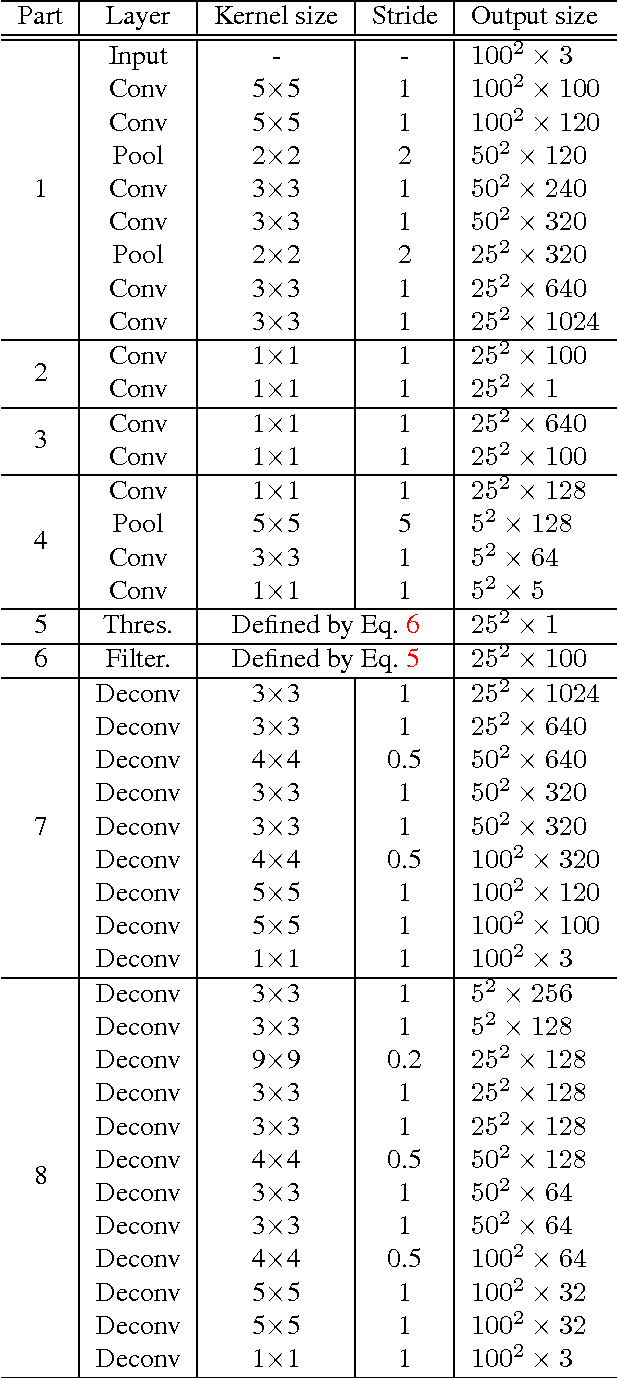
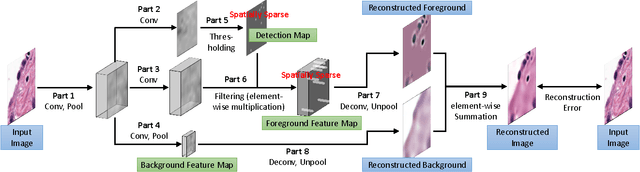
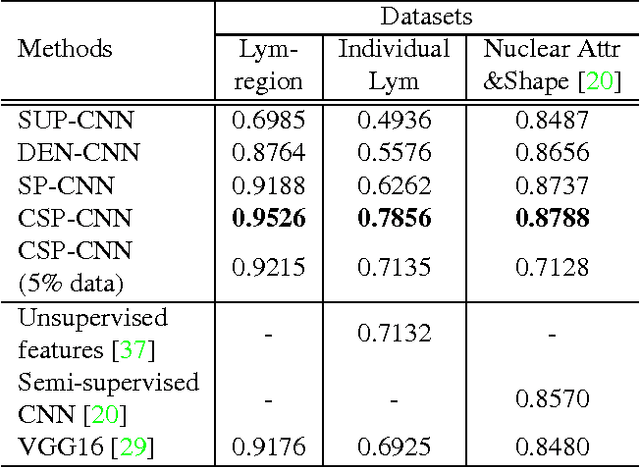
Histopathology images are crucial to the study of complex diseases such as cancer. The histologic characteristics of nuclei play a key role in disease diagnosis, prognosis and analysis. In this work, we propose a sparse Convolutional Autoencoder (CAE) for fully unsupervised, simultaneous nucleus detection and feature extraction in histopathology tissue images. Our CAE detects and encodes nuclei in image patches in tissue images into sparse feature maps that encode both the location and appearance of nuclei. Our CAE is the first unsupervised detection network for computer vision applications. The pretrained nucleus detection and feature extraction modules in our CAE can be fine-tuned for supervised learning in an end-to-end fashion. We evaluate our method on four datasets and reduce the errors of state-of-the-art methods up to 42%. We are able to achieve comparable performance with only 5% of the fully-supervised annotation cost.
Center-Focusing Multi-task CNN with Injected Features for Classification of Glioma Nuclear Images
Jan 10, 2017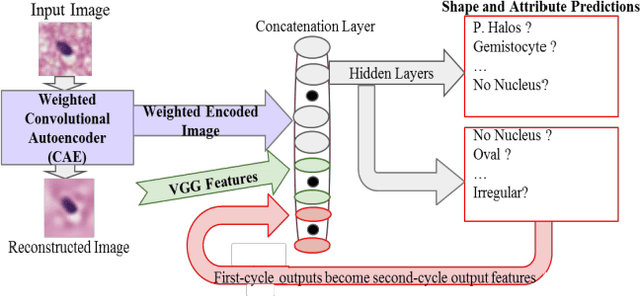
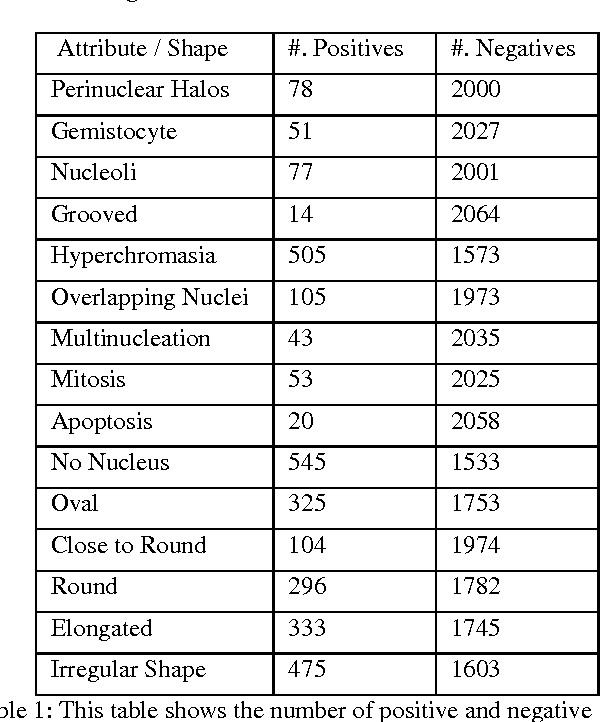
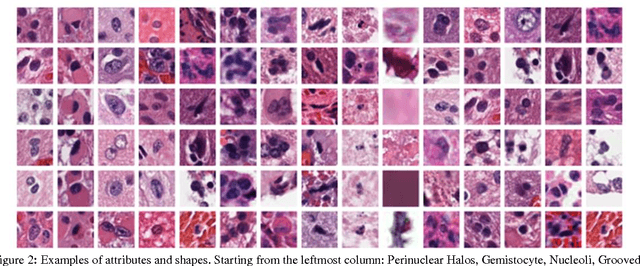
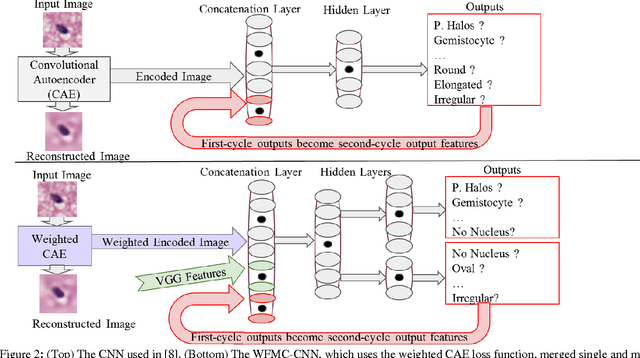
Classifying the various shapes and attributes of a glioma cell nucleus is crucial for diagnosis and understanding the disease. We investigate automated classification of glioma nuclear shapes and visual attributes using Convolutional Neural Networks (CNNs) on pathology images of automatically segmented nuclei. We propose three methods that improve the performance of a previously-developed semi-supervised CNN. First, we propose a method that allows the CNN to focus on the most important part of an image- the image's center containing the nucleus. Second, we inject (concatenate) pre-extracted VGG features into an intermediate layer of our Semi-Supervised CNN so that during training, the CNN can learn a set of complementary features. Third, we separate the losses of the two groups of target classes (nuclear shapes and attributes) into a single-label loss and a multi-label loss so that the prior knowledge of inter-label exclusiveness can be incorporated. On a dataset of 2078 images, the proposed methods combined reduce the error rate of attribute and shape classification by 21.54% and 15.07% respectively compared to the existing state-of-the-art method on the same dataset.