 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSynCLay: Interactive Synthesis of Histology Images from Bespoke Cellular Layouts
Dec 28, 2022



Automated synthesis of histology images has several potential applications in computational pathology. However, no existing method can generate realistic tissue images with a bespoke cellular layout or user-defined histology parameters. In this work, we propose a novel framework called SynCLay (Synthesis from Cellular Layouts) that can construct realistic and high-quality histology images from user-defined cellular layouts along with annotated cellular boundaries. Tissue image generation based on bespoke cellular layouts through the proposed framework allows users to generate different histological patterns from arbitrary topological arrangement of different types of cells. SynCLay generated synthetic images can be helpful in studying the role of different types of cells present in the tumor microenvironmet. Additionally, they can assist in balancing the distribution of cellular counts in tissue images for designing accurate cellular composition predictors by minimizing the effects of data imbalance. We train SynCLay in an adversarial manner and integrate a nuclear segmentation and classification model in its training to refine nuclear structures and generate nuclear masks in conjunction with synthetic images. During inference, we combine the model with another parametric model for generating colon images and associated cellular counts as annotations given the grade of differentiation and cell densities of different cells. We assess the generated images quantitatively and report on feedback from trained pathologists who assigned realism scores to a set of images generated by the framework. The average realism score across all pathologists for synthetic images was as high as that for the real images. We also show that augmenting limited real data with the synthetic data generated by our framework can significantly boost prediction performance of the cellular composition prediction task.
An Aggregation of Aggregation Methods in Computational Pathology
Nov 02, 2022



Image analysis and machine learning algorithms operating on multi-gigapixel whole-slide images (WSIs) often process a large number of tiles (sub-images) and require aggregating predictions from the tiles in order to predict WSI-level labels. In this paper, we present a review of existing literature on various types of aggregation methods with a view to help guide future research in the area of computational pathology (CPath). We propose a general CPath workflow with three pathways that consider multiple levels and types of data and the nature of computation to analyse WSIs for predictive modelling. We categorize aggregation methods according to the context and representation of the data, features of computational modules and CPath use cases. We compare and contrast different methods based on the principle of multiple instance learning, perhaps the most commonly used aggregation method, covering a wide range of CPath literature. To provide a fair comparison, we consider a specific WSI-level prediction task and compare various aggregation methods for that task. Finally, we conclude with a list of objectives and desirable attributes of aggregation methods in general, pros and cons of the various approaches, some recommendations and possible future directions.
Embedding Space Augmentation for Weakly Supervised Learning in Whole-Slide Images
Oct 31, 2022



Multiple Instance Learning (MIL) is a widely employed framework for learning on gigapixel whole-slide images (WSIs) from WSI-level annotations. In most MIL based analytical pipelines for WSI-level analysis, the WSIs are often divided into patches and deep features for patches (i.e., patch embeddings) are extracted prior to training to reduce the overall computational cost and cope with the GPUs' limited RAM. To overcome this limitation, we present EmbAugmenter, a data augmentation generative adversarial network (DA-GAN) that can synthesize data augmentations in the embedding space rather than in the pixel space, thereby significantly reducing the computational requirements. Experiments on the SICAPv2 dataset show that our approach outperforms MIL without augmentation and is on par with traditional patch-level augmentation for MIL training while being substantially faster.
Multi-Scale Attention-based Multiple Instance Learning for Classification of Multi-Gigapixel Histology Images
Sep 07, 2022
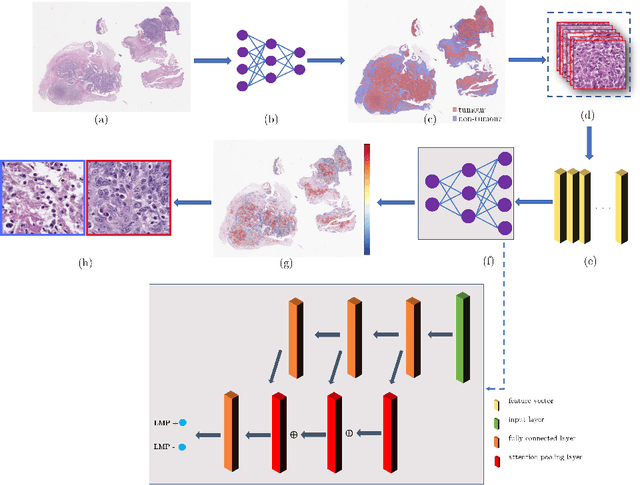
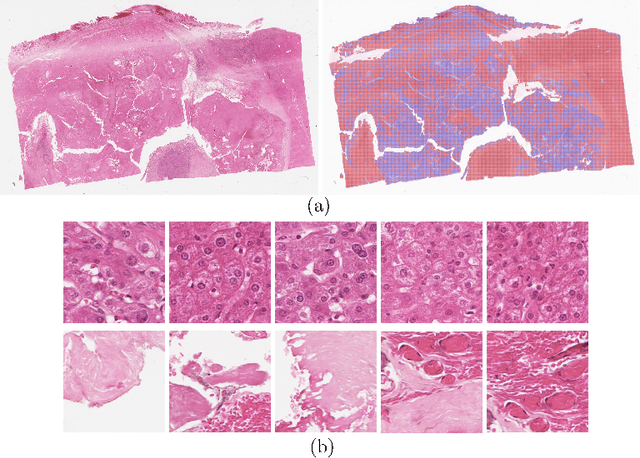
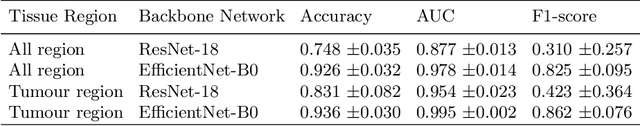
Histology images with multi-gigapixel of resolution yield rich information for cancer diagnosis and prognosis. Most of the time, only slide-level label is available because pixel-wise annotation is labour intensive task. In this paper, we propose a deep learning pipeline for classification in histology images. Using multiple instance learning, we attempt to predict the latent membrane protein 1 (LMP1) status of nasopharyngeal carcinoma (NPC) based on haematoxylin and eosin-stain (H&E) histology images. We utilised attention mechanism with residual connection for our aggregation layers. In our 3-fold cross-validation experiment, we achieved average accuracy, AUC and F1-score 0.936, 0.995 and 0.862, respectively. This method also allows us to examine the model interpretability by visualising attention scores. To the best of our knowledge, this is the first attempt to predict LMP1 status on NPC using deep learning.
Stain-Robust Mitotic Figure Detection for MIDOG 2022 Challenge
Aug 26, 2022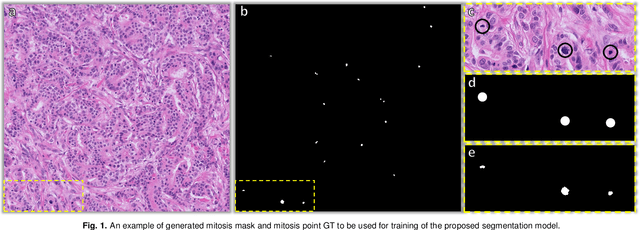
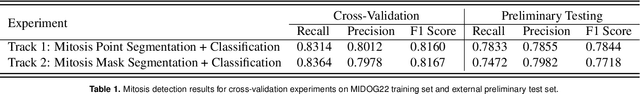
The detection of mitotic figures from different scanners/sites remains an important topic of research, owing to its potential in assisting clinicians with tumour grading. The MItosis DOmain Generalization (MIDOG) 2022 challenge aims to test the robustness of detection models on unseen data from multiple scanners and tissue types for this task. We present a short summary of the approach employed by the TIA Centre team to address this challenge. Our approach is based on a hybrid detection model, where mitotic candidates are segmented, before being refined by a deep learning classifier. Cross-validation on the training images achieved the F1-score of 0.816 and 0.784 on the preliminary test set, demonstrating the generalizability of our model to unseen data from new scanners.
IMPaSh: A Novel Domain-shift Resistant Representation for Colorectal Cancer Tissue Classification
Aug 23, 2022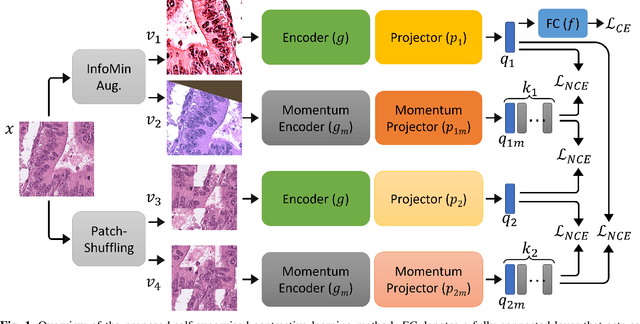
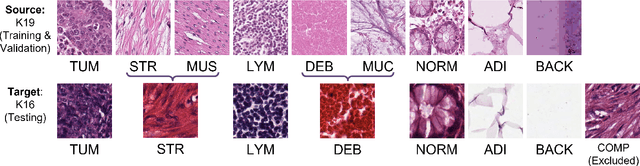
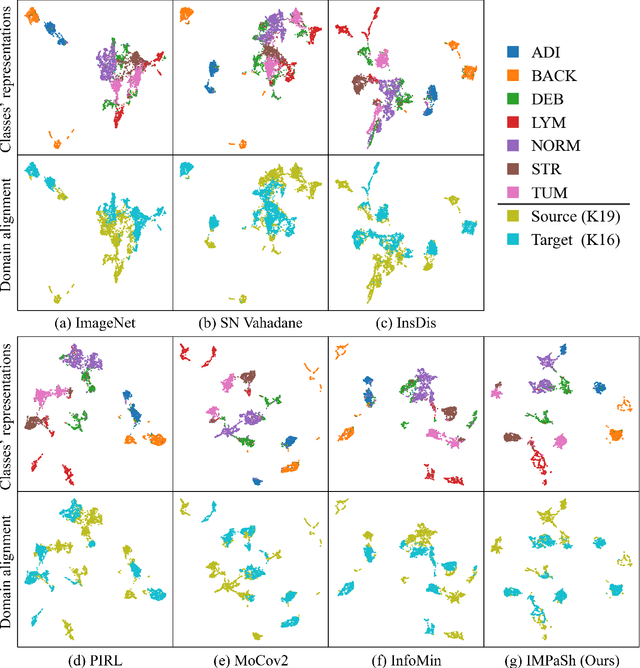
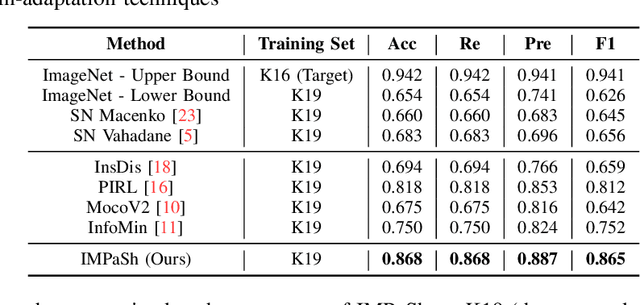
The appearance of histopathology images depends on tissue type, staining and digitization procedure. These vary from source to source and are the potential causes for domain-shift problems. Owing to this problem, despite the great success of deep learning models in computational pathology, a model trained on a specific domain may still perform sub-optimally when we apply them to another domain. To overcome this, we propose a new augmentation called PatchShuffling and a novel self-supervised contrastive learning framework named IMPaSh for pre-training deep learning models. Using these, we obtained a ResNet50 encoder that can extract image representation resistant to domain-shift. We compared our derived representation against those acquired based on other domain-generalization techniques by using them for the cross-domain classification of colorectal tissue images. We show that the proposed method outperforms other traditional histology domain-adaptation and state-of-the-art self-supervised learning methods. Code is available at: https://github.com/trinhvg/IMPash .
TIAger: Tumor-Infiltrating Lymphocyte Scoring in Breast Cancer for the TiGER Challenge
Jun 23, 2022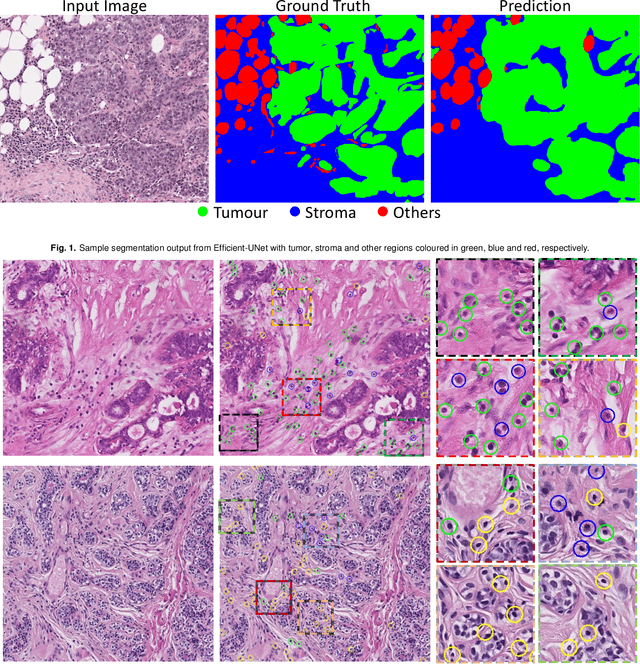
The quantification of tumor-infiltrating lymphocytes (TILs) has been shown to be an independent predictor for prognosis of breast cancer patients. Typically, pathologists give an estimate of the proportion of the stromal region that contains TILs to obtain a TILs score. The Tumor InfiltratinG lymphocytes in breast cancER (TiGER) challenge, aims to assess the prognostic significance of computer-generated TILs scores for predicting survival as part of a Cox proportional hazards model. For this challenge, as the TIAger team, we have developed an algorithm to first segment tumor vs. stroma, before localising the tumor bulk region for TILs detection. Finally, we use these outputs to generate a TILs score for each case. On preliminary testing, our approach achieved a tumor-stroma weighted Dice score of 0.791 and a FROC score of 0.572 for lymphocytic detection. For predicting survival, our model achieved a C-index of 0.719. These results achieved first place across the preliminary testing leaderboards of the TiGER challenge.
Rank the triplets: A ranking-based multiple instance learning framework for detecting HPV infection in head and neck cancers using routine H&E images
Jun 16, 2022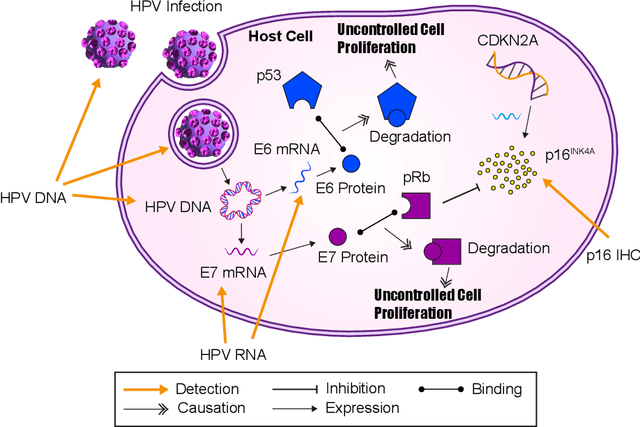
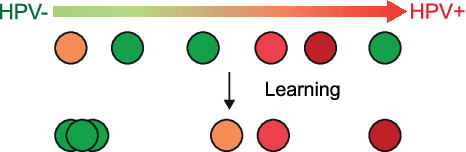

The aetiology of head and neck squamous cell carcinoma (HNSCC) involves multiple carcinogens such as alcohol, tobacco and infection with human papillomavirus (HPV). As the HPV infection influences the prognosis, treatment and survival of patients with HNSCC, it is important to determine the HPV status of these tumours. In this paper, we propose a novel triplet-ranking loss function and a multiple instance learning pipeline for HPV status prediction. This achieves a new state-of-the-art performance in HPV detection using only the routine H&E stained WSIs on two HNSCC cohorts. Furthermore, a comprehensive tumour microenvironment profiling was performed, which characterised the unique patterns between HPV+/- HNSCC from genomic, immunology and cellular perspectives. Positive correlations of the proposed score with different subtypes of T cells (e.g. T cells follicular helper, CD8+ T cells), and negative correlations with macrophages and connective cells (e.g. fibroblast) were identified, which is in line with clinical findings. Unique gene expression profiles were also identified with respect to HPV infection status, and is in line with existing findings.
Metrics reloaded: Pitfalls and recommendations for image analysis validation
Jun 03, 2022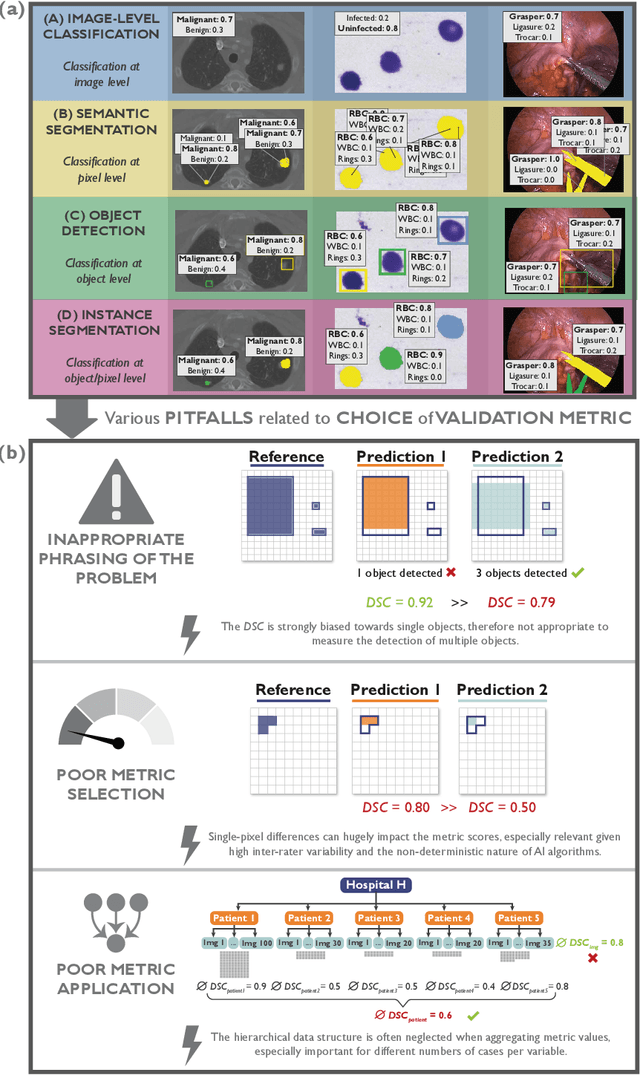
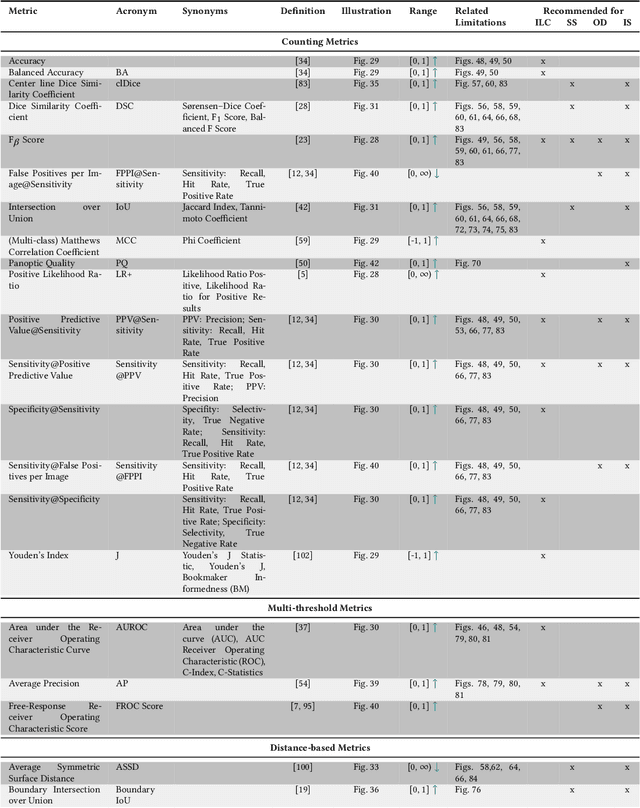
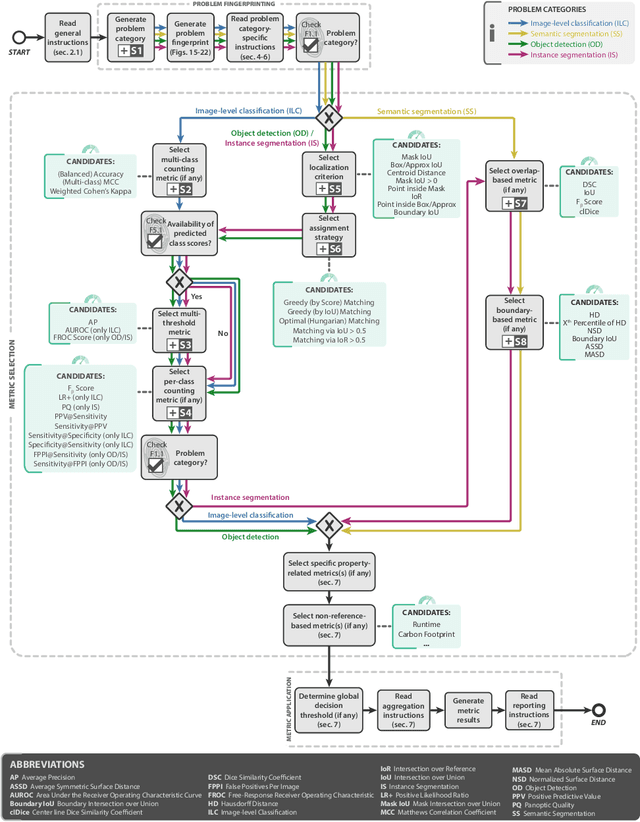
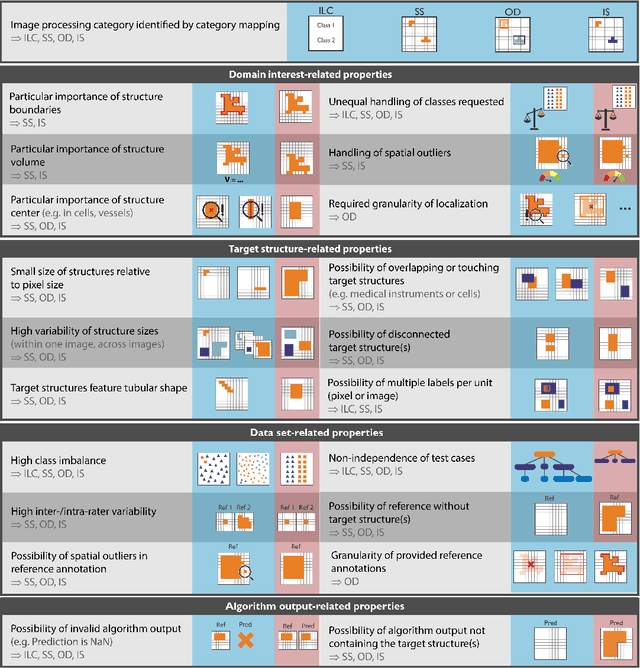
The field of automatic biomedical image analysis crucially depends on robust and meaningful performance metrics for algorithm validation. Current metric usage, however, is often ill-informed and does not reflect the underlying domain interest. Here, we present a comprehensive framework that guides researchers towards choosing performance metrics in a problem-aware manner. Specifically, we focus on biomedical image analysis problems that can be interpreted as a classification task at image, object or pixel level. The framework first compiles domain interest-, target structure-, data set- and algorithm output-related properties of a given problem into a problem fingerprint, while also mapping it to the appropriate problem category, namely image-level classification, semantic segmentation, instance segmentation, or object detection. It then guides users through the process of selecting and applying a set of appropriate validation metrics while making them aware of potential pitfalls related to individual choices. In this paper, we describe the current status of the Metrics Reloaded recommendation framework, with the goal of obtaining constructive feedback from the image analysis community. The current version has been developed within an international consortium of more than 60 image analysis experts and will be made openly available as a user-friendly toolkit after community-driven optimization.
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022
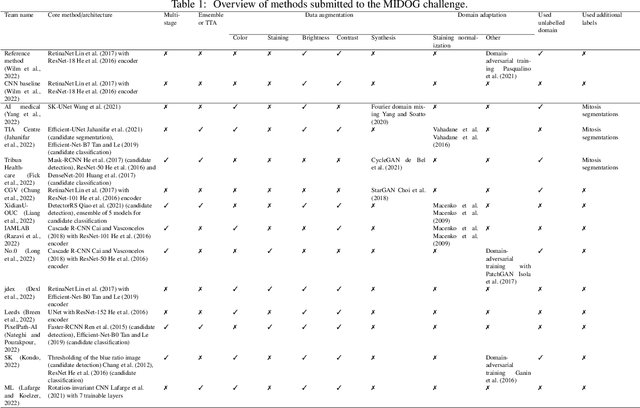
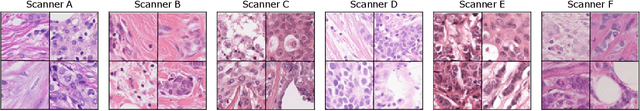
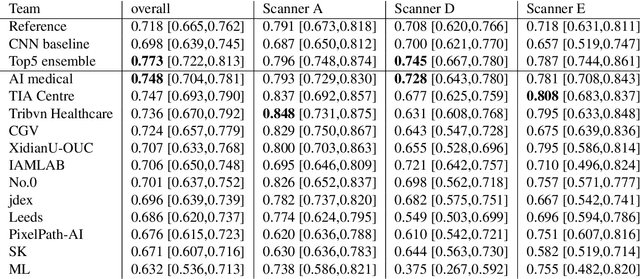
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.