 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMessage-Passing Monte Carlo: Generating low-discrepancy point sets via Graph Neural Networks
May 23, 2024



Discrepancy is a well-known measure for the irregularity of the distribution of a point set. Point sets with small discrepancy are called low-discrepancy and are known to efficiently fill the space in a uniform manner. Low-discrepancy points play a central role in many problems in science and engineering, including numerical integration, computer vision, machine perception, computer graphics, machine learning, and simulation. In this work, we present the first machine learning approach to generate a new class of low-discrepancy point sets named Message-Passing Monte Carlo (MPMC) points. Motivated by the geometric nature of generating low-discrepancy point sets, we leverage tools from Geometric Deep Learning and base our model on Graph Neural Networks. We further provide an extension of our framework to higher dimensions, which flexibly allows the generation of custom-made points that emphasize the uniformity in specific dimensions that are primarily important for the particular problem at hand. Finally, we demonstrate that our proposed model achieves state-of-the-art performance superior to previous methods by a significant margin. In fact, MPMC points are empirically shown to be either optimal or near-optimal with respect to the discrepancy for every dimension and the number of points for which the optimal discrepancy can be determined.
Advancing Graph Convolutional Networks via General Spectral Wavelets
May 22, 2024



Spectral graph convolution, an important tool of data filtering on graphs, relies on two essential decisions; selecting spectral bases for signal transformation and parameterizing the kernel for frequency analysis. While recent techniques mainly focus on standard Fourier transform and vector-valued spectral functions, they fall short in flexibility to describe specific signal distribution for each node, and expressivity of spectral function. In this paper, we present a novel wavelet-based graph convolution network, namely WaveGC, which integrates multi-resolution spectral bases and a matrix-valued filter kernel. Theoretically, we establish that WaveGC can effectively capture and decouple short-range and long-range information, providing superior filtering flexibility, surpassing existing graph convolutional networks and graph Transformers (GTs). To instantiate WaveGC, we introduce a novel technique for learning general graph wavelets by separately combining odd and even terms of Chebyshev polynomials. This approach strictly satisfies wavelet admissibility criteria. Our numerical experiments showcase the capabilities of the new network. By replacing the Transformer part in existing architectures with WaveGC, we consistently observe improvements in both short-range and long-range tasks. This underscores the effectiveness of the proposed model in handling different scenarios. Our code is available at https://github.com/liun-online/WaveGC.
Revealing Decurve Flows for Generalized Graph Propagation
Feb 13, 2024

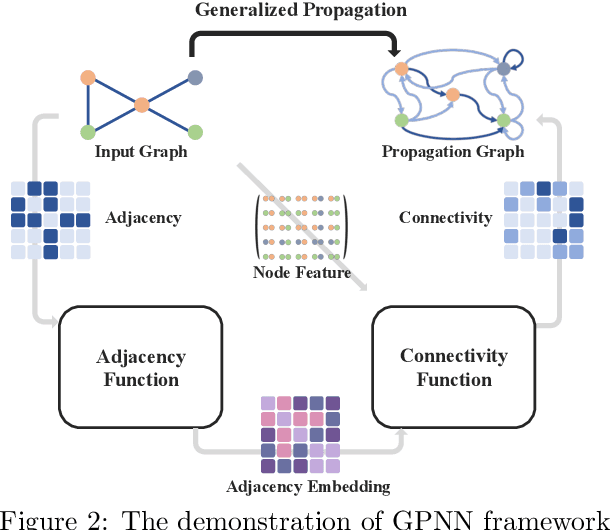

This study addresses the limitations of the traditional analysis of message-passing, central to graph learning, by defining {\em \textbf{generalized propagation}} with directed and weighted graphs. The significance manifest in two ways. \textbf{Firstly}, we propose {\em Generalized Propagation Neural Networks} (\textbf{GPNNs}), a framework that unifies most propagation-based graph neural networks. By generating directed-weighted propagation graphs with adjacency function and connectivity function, GPNNs offer enhanced insights into attention mechanisms across various graph models. We delve into the trade-offs within the design space with empirical experiments and emphasize the crucial role of the adjacency function for model expressivity via theoretical analysis. \textbf{Secondly}, we propose the {\em Continuous Unified Ricci Curvature} (\textbf{CURC}), an extension of celebrated {\em Ollivier-Ricci Curvature} for directed and weighted graphs. Theoretically, we demonstrate that CURC possesses continuity, scale invariance, and a lower bound connection with the Dirichlet isoperimetric constant validating bottleneck analysis for GPNNs. We include a preliminary exploration of learned propagation patterns in datasets, a first in the field. We observe an intriguing ``{\em \textbf{decurve flow}}'' - a curvature reduction during training for models with learnable propagation, revealing the evolution of propagation over time and a deeper connection to over-smoothing and bottleneck trade-off.
Link Prediction with Relational Hypergraphs
Feb 06, 2024Link prediction with knowledge graphs has been thoroughly studied in graph machine learning, leading to a rich landscape of graph neural network architectures with successful applications. Nonetheless, it remains challenging to transfer the success of these architectures to link prediction with relational hypergraphs. The presence of relational hyperedges makes link prediction a task between $k$ nodes for varying choices of $k$, which is substantially harder than link prediction with knowledge graphs, where every relation is binary ($k=2$). In this paper, we propose two frameworks for link prediction with relational hypergraphs and conduct a thorough analysis of the expressive power of the resulting model architectures via corresponding relational Weisfeiler-Leman algorithms, and also via some natural logical formalisms. Through extensive empirical analysis, we validate the power of the proposed model architectures on various relational hypergraph benchmarks. The resulting model architectures substantially outperform every baseline for inductive link prediction, and lead to state-of-the-art results for transductive link prediction. Our study therefore unlocks applications of graph neural networks to fully relational structures.
Setting the Record Straight on Transformer Oversmoothing
Jan 09, 2024Transformer-based models have recently become wildly successful across a diverse set of domains. At the same time, recent work has shown that Transformers are inherently low-pass filters that gradually oversmooth the inputs, reducing the expressivity of their representations. A natural question is: How can Transformers achieve these successes given this shortcoming? In this work we show that in fact Transformers are not inherently low-pass filters. Instead, whether Transformers oversmooth or not depends on the eigenspectrum of their update equations. Our analysis extends prior work in oversmoothing and in the closely-related phenomenon of rank collapse. We show that many successful Transformer models have attention and weights which satisfy conditions that avoid oversmoothing. Based on this analysis, we derive a simple way to parameterize the weights of the Transformer update equations that allows for control over its spectrum, ensuring that oversmoothing does not occur. Compared to a recent solution for oversmoothing, our approach improves generalization, even when training with more layers, fewer datapoints, and data that is corrupted.
How does over-squashing affect the power of GNNs?
Jun 06, 2023Graph Neural Networks (GNNs) are the state-of-the-art model for machine learning on graph-structured data. The most popular class of GNNs operate by exchanging information between adjacent nodes, and are known as Message Passing Neural Networks (MPNNs). Given their widespread use, understanding the expressive power of MPNNs is a key question. However, existing results typically consider settings with uninformative node features. In this paper, we provide a rigorous analysis to determine which function classes of node features can be learned by an MPNN of a given capacity. We do so by measuring the level of pairwise interactions between nodes that MPNNs allow for. This measure provides a novel quantitative characterization of the so-called over-squashing effect, which is observed to occur when a large volume of messages is aggregated into fixed-size vectors. Using our measure, we prove that, to guarantee sufficient communication between pairs of nodes, the capacity of the MPNN must be large enough, depending on properties of the input graph structure, such as commute times. For many relevant scenarios, our analysis results in impossibility statements in practice, showing that over-squashing hinders the expressive power of MPNNs. We validate our theoretical findings through extensive controlled experiments and ablation studies.
A Survey on Oversmoothing in Graph Neural Networks
Mar 20, 2023


Node features of graph neural networks (GNNs) tend to become more similar with the increase of the network depth. This effect is known as over-smoothing, which we axiomatically define as the exponential convergence of suitable similarity measures on the node features. Our definition unifies previous approaches and gives rise to new quantitative measures of over-smoothing. Moreover, we empirically demonstrate this behavior for several over-smoothing measures on different graphs (small-, medium-, and large-scale). We also review several approaches for mitigating over-smoothing and empirically test their effectiveness on real-world graph datasets. Through illustrative examples, we demonstrate that mitigating over-smoothing is a necessary but not sufficient condition for building deep GNNs that are expressive on a wide range of graph learning tasks. Finally, we extend our definition of over-smoothing to the rapidly emerging field of continuous-time GNNs.
Graph Neural Networks for Link Prediction with Subgraph Sketching
Oct 03, 2022
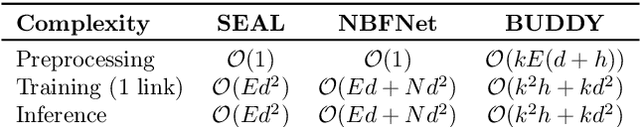
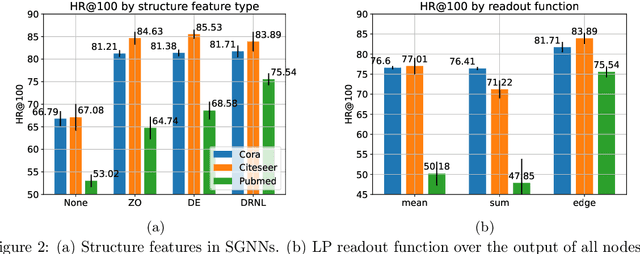
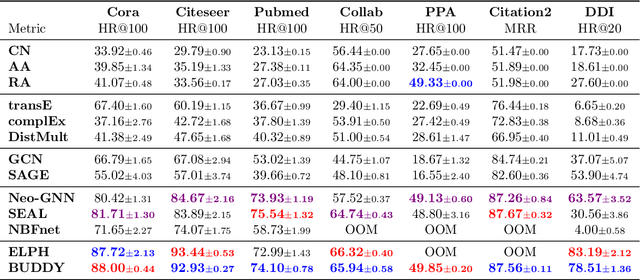
Many Graph Neural Networks (GNNs) perform poorly compared to simple heuristics on Link Prediction (LP) tasks. This is due to limitations in expressive power such as the inability to count triangles (the backbone of most LP heuristics) and because they can not distinguish automorphic nodes (those having identical structural roles). Both expressiveness issues can be alleviated by learning link (rather than node) representations and incorporating structural features such as triangle counts. Since explicit link representations are often prohibitively expensive, recent works resorted to subgraph-based methods, which have achieved state-of-the-art performance for LP, but suffer from poor efficiency due to high levels of redundancy between subgraphs. We analyze the components of subgraph GNN (SGNN) methods for link prediction. Based on our analysis, we propose a novel full-graph GNN called ELPH (Efficient Link Prediction with Hashing) that passes subgraph sketches as messages to approximate the key components of SGNNs without explicit subgraph construction. ELPH is provably more expressive than Message Passing GNNs (MPNNs). It outperforms existing SGNN models on many standard LP benchmarks while being orders of magnitude faster. However, it shares the common GNN limitation that it is only efficient when the dataset fits in GPU memory. Accordingly, we develop a highly scalable model, called BUDDY, which uses feature precomputation to circumvent this limitation without sacrificing predictive performance. Our experiments show that BUDDY also outperforms SGNNs on standard LP benchmarks while being highly scalable and faster than ELPH.
Gradient Gating for Deep Multi-Rate Learning on Graphs
Oct 02, 2022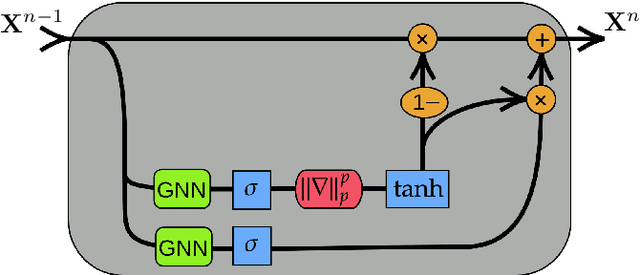
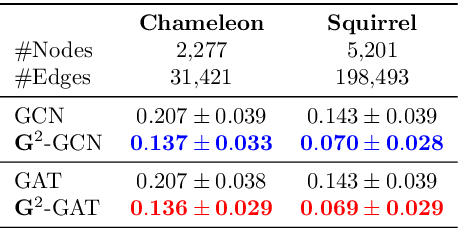
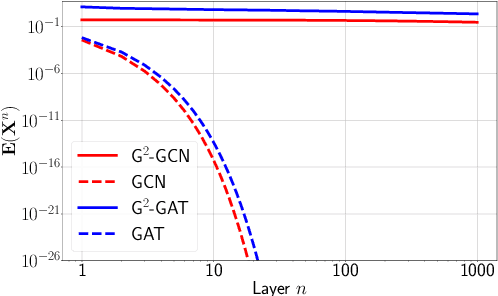
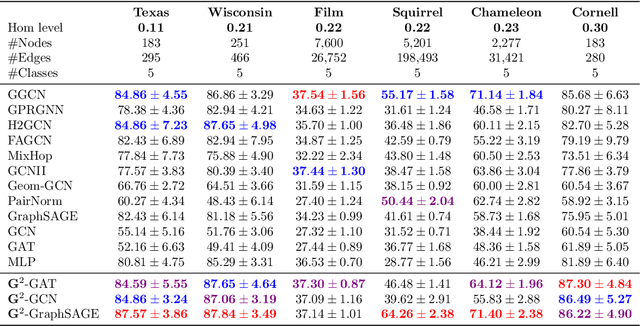
We present Gradient Gating (G$^2$), a novel framework for improving the performance of Graph Neural Networks (GNNs). Our framework is based on gating the output of GNN layers with a mechanism for multi-rate flow of message passing information across nodes of the underlying graph. Local gradients are harnessed to further modulate message passing updates. Our framework flexibly allows one to use any basic GNN layer as a wrapper around which the multi-rate gradient gating mechanism is built. We rigorously prove that G$^2$ alleviates the oversmoothing problem and allows the design of deep GNNs. Empirical results are presented to demonstrate that the proposed framework achieves state-of-the-art performance on a variety of graph learning tasks, including on large-scale heterophilic graphs.
Understanding and Extending Subgraph GNNs by Rethinking Their Symmetries
Jun 22, 2022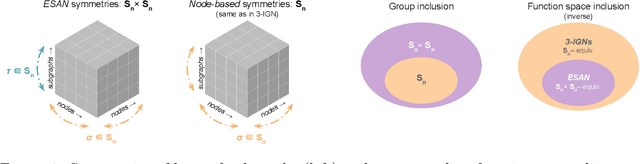
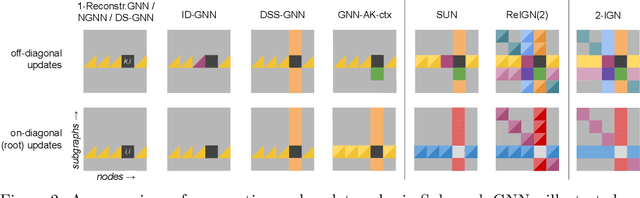
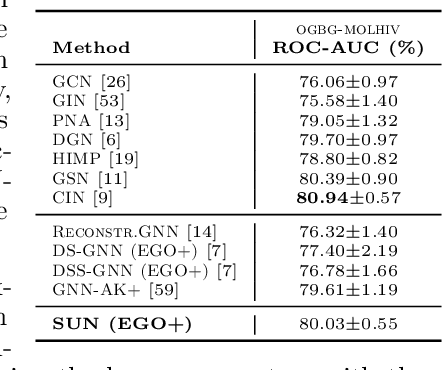
Subgraph GNNs are a recent class of expressive Graph Neural Networks (GNNs) which model graphs as collections of subgraphs. So far, the design space of possible Subgraph GNN architectures as well as their basic theoretical properties are still largely unexplored. In this paper, we study the most prominent form of subgraph methods, which employs node-based subgraph selection policies such as ego-networks or node marking and deletion. We address two central questions: (1) What is the upper-bound of the expressive power of these methods? and (2) What is the family of equivariant message passing layers on these sets of subgraphs?. Our first step in answering these questions is a novel symmetry analysis which shows that modelling the symmetries of node-based subgraph collections requires a significantly smaller symmetry group than the one adopted in previous works. This analysis is then used to establish a link between Subgraph GNNs and Invariant Graph Networks (IGNs). We answer the questions above by first bounding the expressive power of subgraph methods by 3-WL, and then proposing a general family of message-passing layers for subgraph methods that generalises all previous node-based Subgraph GNNs. Finally, we design a novel Subgraph GNN dubbed SUN, which theoretically unifies previous architectures while providing better empirical performance on multiple benchmarks.