 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTraining on test proteins improves fitness, structure, and function prediction
Nov 04, 2024



Data scarcity and distribution shifts often hinder the ability of machine learning models to generalize when applied to proteins and other biological data. Self-supervised pre-training on large datasets is a common method to enhance generalization. However, striving to perform well on all possible proteins can limit model's capacity to excel on any specific one, even though practitioners are often most interested in accurate predictions for the individual protein they study. To address this limitation, we propose an orthogonal approach to achieve generalization. Building on the prevalence of self-supervised pre-training, we introduce a method for self-supervised fine-tuning at test time, allowing models to adapt to the test protein of interest on the fly and without requiring any additional data. We study our test-time training (TTT) method through the lens of perplexity minimization and show that it consistently enhances generalization across different models, their scales, and datasets. Notably, our method leads to new state-of-the-art results on the standard benchmark for protein fitness prediction, improves protein structure prediction for challenging targets, and enhances function prediction accuracy.
Think While You Generate: Discrete Diffusion with Planned Denoising
Oct 08, 2024



Discrete diffusion has achieved state-of-the-art performance, outperforming or approaching autoregressive models on standard benchmarks. In this work, we introduce Discrete Diffusion with Planned Denoising (DDPD), a novel framework that separates the generation process into two models: a planner and a denoiser. At inference time, the planner selects which positions to denoise next by identifying the most corrupted positions in need of denoising, including both initially corrupted and those requiring additional refinement. This plan-and-denoise approach enables more efficient reconstruction during generation by iteratively identifying and denoising corruptions in the optimal order. DDPD outperforms traditional denoiser-only mask diffusion methods, achieving superior results on language modeling benchmarks such as text8, OpenWebText, and token-based generation on ImageNet $256 \times 256$. Notably, in language modeling, DDPD significantly reduces the performance gap between diffusion-based and autoregressive methods in terms of generative perplexity. Code is available at https://github.com/liusulin/DDPD.
Generative Modeling of Molecular Dynamics Trajectories
Sep 26, 2024



Molecular dynamics (MD) is a powerful technique for studying microscopic phenomena, but its computational cost has driven significant interest in the development of deep learning-based surrogate models. We introduce generative modeling of molecular trajectories as a paradigm for learning flexible multi-task surrogate models of MD from data. By conditioning on appropriately chosen frames of the trajectory, we show such generative models can be adapted to diverse tasks such as forward simulation, transition path sampling, and trajectory upsampling. By alternatively conditioning on part of the molecular system and inpainting the rest, we also demonstrate the first steps towards dynamics-conditioned molecular design. We validate the full set of these capabilities on tetrapeptide simulations and show that our model can produce reasonable ensembles of protein monomers. Altogether, our work illustrates how generative modeling can unlock value from MD data towards diverse downstream tasks that are not straightforward to address with existing methods or even MD itself. Code is available at https://github.com/bjing2016/mdgen.
Transition Path Sampling with Boltzmann Generator-based MCMC Moves
Dec 08, 2023



Sampling all possible transition paths between two 3D states of a molecular system has various applications ranging from catalyst design to drug discovery. Current approaches to sample transition paths use Markov chain Monte Carlo and rely on time-intensive molecular dynamics simulations to find new paths. Our approach operates in the latent space of a normalizing flow that maps from the molecule's Boltzmann distribution to a Gaussian, where we propose new paths without requiring molecular simulations. Using alanine dipeptide, we explore Metropolis-Hastings acceptance criteria in the latent space for exact sampling and investigate different latent proposal mechanisms.
Harmonic Self-Conditioned Flow Matching for Multi-Ligand Docking and Binding Site Design
Oct 09, 2023



A significant amount of protein function requires binding small molecules, including enzymatic catalysis. As such, designing binding pockets for small molecules has several impactful applications ranging from drug synthesis to energy storage. Towards this goal, we first develop HarmonicFlow, an improved generative process over 3D protein-ligand binding structures based on our self-conditioned flow matching objective. FlowSite extends this flow model to jointly generate a protein pocket's discrete residue types and the molecule's binding 3D structure. We show that HarmonicFlow improves upon the state-of-the-art generative processes for docking in simplicity, generality, and performance. Enabled by this structure modeling, FlowSite designs binding sites substantially better than baseline approaches and provides the first general solution for binding site design.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023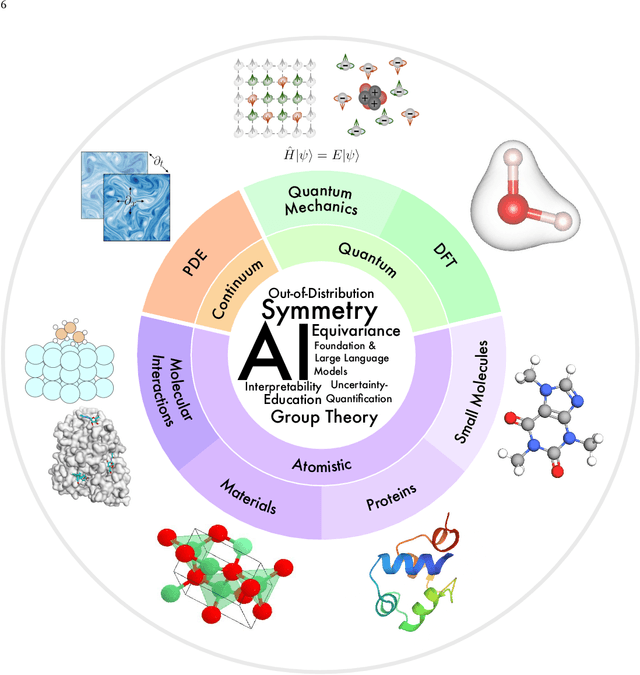



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
DiffDock-PP: Rigid Protein-Protein Docking with Diffusion Models
Apr 08, 2023



Understanding how proteins structurally interact is crucial to modern biology, with applications in drug discovery and protein design. Recent machine learning methods have formulated protein-small molecule docking as a generative problem with significant performance boosts over both traditional and deep learning baselines. In this work, we propose a similar approach for rigid protein-protein docking: DiffDock-PP is a diffusion generative model that learns to translate and rotate unbound protein structures into their bound conformations. We achieve state-of-the-art performance on DIPS with a median C-RMSD of 4.85, outperforming all considered baselines. Additionally, DiffDock-PP is faster than all search-based methods and generates reliable confidence estimates for its predictions. Our code is publicly available at $\texttt{https://github.com/ketatam/DiffDock-PP}$
Task-Agnostic Graph Neural Network Evaluation via Adversarial Collaboration
Jan 27, 2023It has been increasingly demanding to develop reliable Graph Neural Network (GNN) evaluation methods to quantify the progress of the rapidly expanding GNN research. Existing GNN benchmarking methods focus on comparing the GNNs with respect to their performances on some node/graph classification/regression tasks in certain datasets. There lacks a principled, task-agnostic method to directly compare two GNNs. Moreover, most of the existing graph self-supervised learning (SSL) works incorporate handcrafted augmentations to the graph, which has several severe difficulties due to the unique characteristics of graph-structured data. To address the aforementioned issues, we propose GraphAC (Graph Adversarial Collaboration) -- a conceptually novel, principled, task-agnostic, and stable framework for evaluating GNNs through contrastive self-supervision. GraphAC succeeds in distinguishing GNNs of different expressiveness across various aspects, and has been proven to be a principled and reliable GNN evaluation method, eliminating the need for handcrafted augmentations for stable SSL.
Generalized Laplacian Positional Encoding for Graph Representation Learning
Nov 10, 2022



Graph neural networks (GNNs) are the primary tool for processing graph-structured data. Unfortunately, the most commonly used GNNs, called Message Passing Neural Networks (MPNNs) suffer from several fundamental limitations. To overcome these limitations, recent works have adapted the idea of positional encodings to graph data. This paper draws inspiration from the recent success of Laplacian-based positional encoding and defines a novel family of positional encoding schemes for graphs. We accomplish this by generalizing the optimization problem that defines the Laplace embedding to more general dissimilarity functions rather than the 2-norm used in the original formulation. This family of positional encodings is then instantiated by considering p-norms. We discuss a method for calculating these positional encoding schemes, implement it in PyTorch and demonstrate how the resulting positional encoding captures different properties of the graph. Furthermore, we demonstrate that this novel family of positional encodings can improve the expressive power of MPNNs. Lastly, we present preliminary experimental results.
Equivariant 3D-Conditional Diffusion Models for Molecular Linker Design
Oct 11, 2022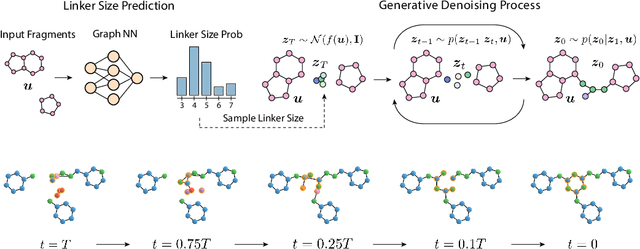
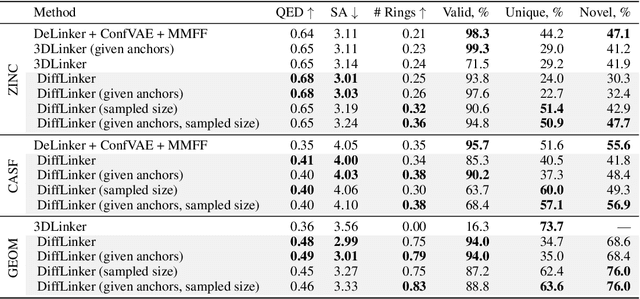
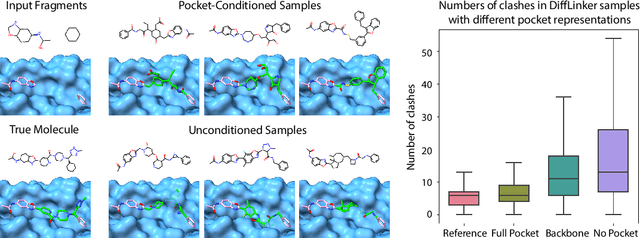
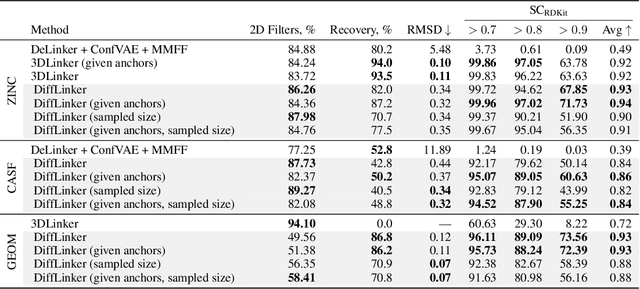
Fragment-based drug discovery has been an effective paradigm in early-stage drug development. An open challenge in this area is designing linkers between disconnected molecular fragments of interest to obtain chemically-relevant candidate drug molecules. In this work, we propose DiffLinker, an E(3)-equivariant 3D-conditional diffusion model for molecular linker design. Given a set of disconnected fragments, our model places missing atoms in between and designs a molecule incorporating all the initial fragments. Unlike previous approaches that are only able to connect pairs of molecular fragments, our method can link an arbitrary number of fragments. Additionally, the model automatically determines the number of atoms in the linker and its attachment points to the input fragments. We demonstrate that DiffLinker outperforms other methods on the standard datasets generating more diverse and synthetically-accessible molecules. Besides, we experimentally test our method in real-world applications, showing that it can successfully generate valid linkers conditioned on target protein pockets.