 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRECOVER: sequential model optimization platform for combination drug repurposing identifies novel synergistic compounds in vitro
Feb 07, 2022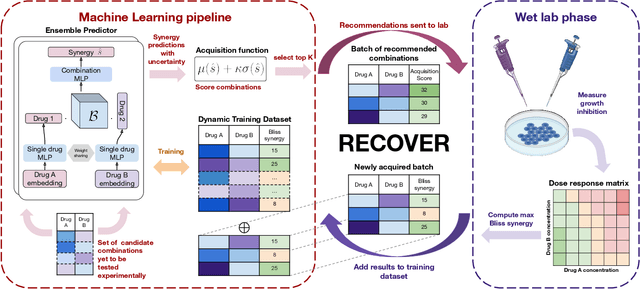

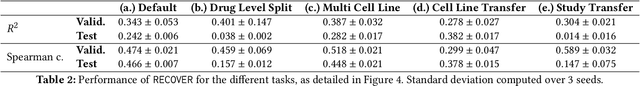
Selecting optimal drug repurposing combinations for further preclinical development is a challenging technical feat. Due to the toxicity of many therapeutic agents (e.g., chemotherapy), practitioners have favoured selection of synergistic compounds whereby lower doses can be used whilst maintaining high efficacy. For a fixed small molecule library, an exhaustive combinatorial chemical screen becomes infeasible to perform for academic and industry laboratories alike. Deep learning models have achieved state-of-the-art results in silico for the prediction of synergy scores. However, databases of drug combinations are highly biased towards synergistic agents and these results do not necessarily generalise out of distribution. We employ a sequential model optimization search applied to a deep learning model to quickly discover highly synergistic drug combinations active against a cancer cell line, while requiring substantially less screening than an exhaustive evaluation. Through iteratively adapting the model to newly acquired data, after only 3 rounds of ML-guided experimentation (including a calibration round), we find that the set of combinations queried by our model is enriched for highly synergistic combinations. Remarkably, we rediscovered a synergistic drug combination that was later confirmed to be under study within clinical trials.
Utilising Graph Machine Learning within Drug Discovery and Development
Dec 09, 2020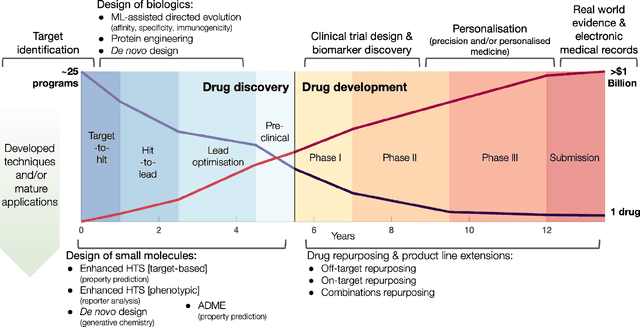
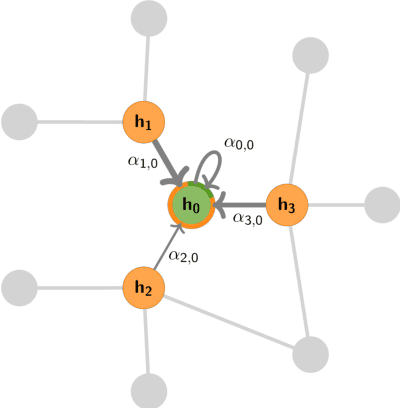
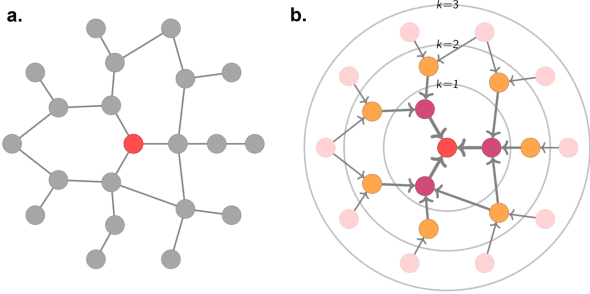
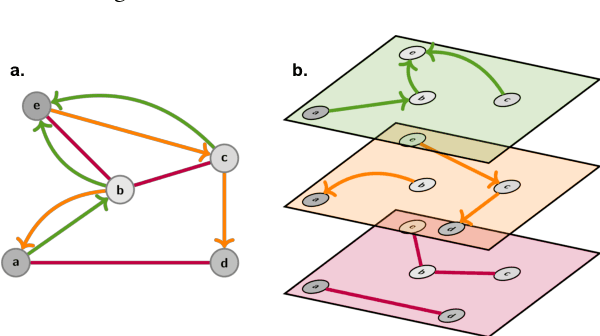
Graph Machine Learning (GML) is receiving growing interest within the pharmaceutical and biotechnology industries for its ability to model biomolecular structures, the functional relationships between them, and integrate multi-omic datasets - amongst other data types. Herein, we present a multidisciplinary academic-industrial review of the topic within the context of drug discovery and development. After introducing key terms and modelling approaches, we move chronologically through the drug development pipeline to identify and summarise work incorporating: target identification, design of small molecules and biologics, and drug repurposing. Whilst the field is still emerging, key milestones including repurposed drugs entering in vivo studies, suggest graph machine learning will become a modelling framework of choice within biomedical machine learning.